Susilo Hariyanto*![]() | Siti Khabibah
| Siti Khabibah![]() | Retno Putri Dwi Rahmawati
| Retno Putri Dwi Rahmawati![]() | Bibit Waluyo Aji
| Bibit Waluyo Aji![]()
© 2025 The authors. This article is published by IIETA and is licensed under the CC BY 4.0 license (http://creativecommons.org/licenses/by/4.0/).
OPEN ACCESS
Accurate prediction of ribonucleic acid (RNA) binding sites is essential for deciphering RNA–protein interactions and understanding post-transcriptional gene regulation. While Graph Convolutional Networks (GCNs) effectively capture complex topological features in biological data, their performance heavily relies on the choice of optimization algorithm. This study proposes FuzzyAdam, a novel gradient-based optimizer that integrates fuzzy logic into the adaptive learning framework of standard Adam to improve convergence behavior in CNN-GCN hybrid models. Unlike Adam, FuzzyAdam dynamically adjusts learning rates based on fuzzy inference over gradient trends, aiming to reduce oscillations and misclassification. To assess its effectiveness, we trained a CNN-GCN architecture on a balanced dataset of 997 image-encoded RNA binding and non-binding sequences. Compared to standard Adam, FuzzyAdam achieved higher performance across all metrics: 98.39% accuracy, 98.39% F1-score, 98.42% precision, and 98.39% recall, with more stable convergence and reduced false negatives as indicated by confusion matrix analysis. Although the model does not explicitly model regulatory mechanisms, improved classification of binding sites can facilitate downstream analyses related to post-transcriptional control. FuzzyAdam offers a robust and interpretable optimization strategy, with potential utility for broader bioinformatics tasks involving graph-based or structurally encoded inputs.
ribonucleic acid (RNA) binding site, graph convolutional network, hybrid deep learning, FuzzyAdam optimizer, bioinformatics, deep learning
Ribonucleic acid (RNA)-binding proteins (RBPs) are integral to post-transcriptional regulation in eukaryotic cells, orchestrating a wide array of biological processes including RNA splicing, transport, stability, localization, and translation control [1, 2]. By forming ribonucleoprotein complexes (RNPs) through interactions with coding and non-coding RNAs, RBPs establish dynamic regulatory networks that fine-tune gene expression in a context-dependent manner [3, 4]. Disruption in RBP-RNA interactions has been implicated in a variety of pathophysiological conditions, ranging from neurodegenerative disorders like amyotrophic lateral sclerosis (ALS) and frontotemporal dementia to tumorigenesis in various cancers [5, 6].
Conventional experimental methods for RBP identification, such as cross-linking immunoprecipitation followed by sequencing (CLIP-seq) [7, 8], RNA immunoprecipitation sequencing (RIP-seq) [9], and electrophoretic mobility shift assays (EMSAs) [10], have provided foundational insights into RNA–protein interactions. However, these techniques are inherently labor-intensive, low-throughput, and require significant experimental optimization, which limits their scalability in large-scale or dynamic cellular contexts.
In response to these limitations, computational approaches—especially those based on machine learning—have emerged as viable alternatives for predicting RBP binding sites directly from nucleotide sequences. Among these, deep learning methods have shown remarkable promise due to their ability to automatically learn complex patterns from raw biological data without the need for manual feature engineering [11-13]. Specifically, convolutional neural networks (CNNs) have been widely adopted to capture local sequence motifs relevant to RBP binding, as demonstrated by DeepCLIP [14], which combines CNN and long short-term memory (LSTM) layers to model both spatial and contextual dependencies in sequence data.
Building upon the success of CNN-based models, recent studies have explored the incorporation of graph-based architectures such as graph convolutional networks (GCNs), which allow the integration of structural relationships between RNA nucleotides into the prediction framework. For instance, DeepPN [15] combines CNNs and GCNs to simultaneously leverage both local sequence context and the underlying RNA secondary structure, achieving improved predictive performance without explicit structural input.
In addition to these neural models, ensemble learning methods like HydRA [16] and DeepFusion [17] have further extended the scope of RBP prediction by integrating diverse data modalities, including protein sequence features and evolutionary information, to enhance classification accuracy and generalizability. NucleicNet [18], for example, frames RBP prediction as a residue-level classification task, offering fine-grained insights into protein-RNA recognition mechanisms at atomic resolution.
Despite remarkable progress in the development of deep learning models for RNA–protein binding site prediction, these methods often fall short in capturing the full complexity of RNA–protein interactions. Such interactions are governed by a wide array of biochemical, topological, and thermodynamic factors that are difficult to model using standard neural architectures. Moreover, many state-of-the-art models lack interpretability, functioning as “black boxes” that hinder biological validation and limit translational applicability. This poses a significant barrier, especially in biomedical contexts where model explainability is crucial for generating actionable insights.
To address these limitations, recent studies have increasingly explored the integration of fuzzy logic into machine learning and deep learning pipelines. Fuzzy logic has been applied across various machine learning domains [19-21] and has consistently improved model performance by enhancing robustness, convergence, and generalization. Fuzzy logic offers a mathematically grounded framework for handling uncertainty, ambiguity, and data imprecision—characteristics that are highly prevalent in biological datasets. For instance, hybrid neuro-fuzzy systems have demonstrated improved transparency and interpretability in complex models through rule-based reasoning mechanisms without sacrificing predictive performance [22]. Likewise, the intuitionistic fuzzy broad learning system (IF-BLS) has shown superior diagnostic accuracy in noisy datasets such as Alzheimer’s disease classification [23]. Among recent contributions, reference [24] provided a systematic review of predictive uncertainty estimation techniques in machine learning, highlighting the value of probabilistic frameworks such as fuzzy logic for improving both the reliability and robustness of model outcomes under uncertain or imbalanced conditions.
The practical utility of fuzzy logic is further exemplified in clinical applications, where real-time interpretability and robustness are paramount. In stroke rehabilitation systems, fuzzy logic has been instrumental in enabling responsive feedback control, thereby enhancing therapy outcomes in environments characterized by patient variability and uncertainty. For instance, Das et al. [25] introduced a hybrid model combining fuzzy logic with machine learning to monitor lower limb exercises in stroke patients, facilitating real-time feedback and progress tracking without human intervention [25].
In the realm of medical imaging, fuzzy-augmented deep learning frameworks have demonstrated superior performance over traditional architectures by effectively managing ambiguous pixel-level data and improving diagnostic reliability. A comprehensive survey by Zheng et al. [26] highlighted the efficacy of fuzzy deep learning models in handling uncertain medical data, emphasizing their advantages in enhancing model interpretability and generalization across various clinical scenarios. Additionally, a novel ensemble fuzzy deep learning approach was proposed for brain MRI analysis, integrating volumetric fuzzy pooling and attention mechanisms to improve the segmentation of brain tissues and abnormalities, thereby advancing diagnostic accuracy [27].
Motivated by these challenges and limitations, we focus our methodological innovation on the development of a novel fuzzy logic-augmented optimizer, named FuzzyAdam, specifically tailored for RNA–protein binding site prediction in noisy, imbalanced, and temporally dynamic biological datasets. Unlike conventional optimizers such as Adam, FuzzyAdam introduces an adaptive learning mechanism driven by fuzzy inference rules that respond to fluctuations in loss dynamics and gradient behaviour. This approach enhances training stability, improves generalization, and promotes interpretability, without requiring architectural changes or manual reweighting.
Our contribution is centered on the design and implementation of FuzzyAdam, which enhances convergence stability without requiring any modification to the underlying model architecture or data representation. This approach offers a principled mechanism for handling uncertainty during training—a characteristic often presents in biological datasets—even when explicit noise or imbalance is not modeled.
To the best of our knowledge, this is the first application of fuzzy-enhanced optimization within the context of RBP interaction modeling, offering a new dimension of control and interpretability in biological deep learning frameworks.
The Adam (Adaptive Moment Estimation) optimizer is a first-order gradient-based method widely used in deep learning due to its empirical effectiveness, particularly in training large-scale and noisy models. It combines the advantages of two other popular optimization techniques: AdaGrad and RMSProp, by computing adaptive learning rates for each parameter.
Formally in reference [28], let $\theta_t \in \mathbb{R}^d$ denote the parameter vector at iteration t, and $g_t=\nabla_\theta \mathcal{L}\left(\theta_t\right)$ be the stochastic gradient of a loss function $\mathcal{L}$ at step t. Adam maintains exponential moving averages of both the gradients and their squares:
$m_t=\beta_1 m_{t-1}+\left(1-\beta_1\right) g_t$ (1)
$v_t=\beta_2 v_{t-1}+\left(1-\beta_2\right) g_t^2$ (2)
To correct initialization bias, Adam computes bias-corrected moment estimates:
$\hat{m}_t=\frac{m_t}{1-\beta_1^t}, \hat{v}_t=\frac{v_t}{1-\beta_2^t}$ (3)
The parameter update rule is then given by:
$\theta_{t+1}=\theta_t-\frac{\eta}{\sqrt{\hat{v}_t}+\varepsilon} \hat{m}_t$ (4)
where, $\eta$ is the learning rate, $\beta_1, \beta_2 \in[0,1)$ are decay rates for the moment estimates, $\varepsilon \ll 1$ is a small constant to avoid division by zero (typically $\varepsilon=10^{-8}$).
This formulation allows Adam to adaptively scale learning rates for each parameter based on its gradient history, thereby improving optimization stability and convergence speed—especially in problems with sparse gradients or non-stationary objectives.
3.1 Conceptual overview
The FuzzyAdam optimizer is a novel extension of the standard Adam optimizer, incorporating fuzzy logic-based adaptation into the learning rate dynamics. While Adam utilizes first and second moment estimates of gradients to perform parameter updates with adaptive learning rates, FuzzyAdam introduces a fuzzy inference mechanism that dynamically adjusts the effective learning rate scaling at each iteration based on recent training dynamics.
Let $\theta_t$ denote the model parameters at iteration t, and let $\mathcal{L}\left(\theta_t\right)$ be the corresponding loss. The update rule for FuzzyAdam is defined as:
$\theta_{t+1}=\theta_t-\eta \cdot \lambda_t \cdot \frac{\widehat{m}_t}{\sqrt{\hat{v}_t}+\varepsilon}$ (5)
where:
$\lambda_t \in \mathbb{R}^{+}$ is a fuzzy scaling factor adaptively determined at each step through a fuzzy inference system. The fuzzy factor $\lambda_t$ modulates the update magnitude in a context-aware manner by evaluating features extracted from training behavior, such as: Change in loss: $\Delta \mathcal{L}_t=\mathcal{L}_t-\mathcal{L}_{t-1}$ and Gradient norm: $\left\|g_t\right\|$.
The full step-by-step procedure of FuzzyAdam is outlined in Algorithm 1.
|
Algorithm 1: FuzzyAdam Optimizer |
|
Input: Initial parameters $\theta_0$, learning rate $\eta$, exponential decay rates $\beta_1, \beta_2 \in[0,1)$, small constant $\varepsilon>0$, fuzzy momentum coefficient $\gamma \in[0,1]$ Initialize: $m_0 \leftarrow 0$ (first moment vector) $v_0 \leftarrow 0$ (second moment vector) fuzzy_score $\longleftarrow$ 1.0 prev_loss $\longleftarrow$ None for $t=1$ to T do:
$\Delta \mathcal{L}_t \leftarrow \mathcal{L}_t-\mathcal{L}_{t-1}$, g_norm $\leftarrow\left\|g_{\mathrm{t}}\right\|$, v_std $\leftarrow \sqrt{ }\left(\operatorname{mean}\left(v_t\right)\right)$ Apply fuzzy rules: if $\Delta \mathcal{L}>0$: $\lambda \leftarrow 0.5$ if g_norm > 1 else 0.8 else if $\Delta \mathcal{L}<0$: $\lambda \leftarrow 1.05$ if v_std < 0.1 else 1.01 Smooth fuzzy factor: fuzzy $_{\text {score }} \leftarrow \gamma \cdot$ fuzzy$_{\text {score }}+(1-\gamma) \cdot \lambda$ else: fuzzy_score $\longleftarrow$ 1.0
$\theta_{t+1} \leftarrow \theta_t-\eta \cdot$ fuzzy_score $\cdot \widehat{m}_t /$ denom
return $\theta_T$ |
FuzzyAdam extends the standard Adam algorithm by embedding fuzzy reasoning to improve adaptivity and robustness. Unlike traditional optimizers that rely on fixed or heuristically scheduled learning rates, FuzzyAdam dynamically adjusts the effective step size in each iteration. This adjustment is guided by fuzzy inference over signals such as gradient magnitude, loss trajectory, and momentum variance—enabling smoother updates and improved convergence.
By integrating fuzzy logic, FuzzyAdam captures vague and nonlinear dependencies in training dynamics that are otherwise difficult to encode using classical heuristics. As a result, it offers several desirable properties:
In summary, FuzzyAdam can be viewed as a cognitively augmented optimizer that combines the statistical strength of Adam with the flexible reasoning capabilities of fuzzy logic. This synergy makes it particularly effective for scenarios where manual learning rate tuning is impractical, and training stability is critical.
3.2 Convergence analysis
Theorem: Let $\mathcal{L}(\theta)$ be a continuously differentiable loss function that is bounded below and has Lipschitz continuous gradients with constant $\mathcal{L}_g>0$, i.e.,
$\left\|\nabla \mathcal{L}(\theta)-\nabla \mathcal{L}\left(\theta^{\prime}\right)\right\| \leq \mathcal{L}_g\left\|\theta-\theta^{\prime}\right\|$ forall $\theta, \theta^{\prime}$.
Assume:
Then, the sequence $\left\{\theta_t\right\}$ generated by the FuzzyAdam update rule:
$\theta_{t+1}=\theta_t-\eta \cdot \lambda_t \cdot \frac{\widehat{m}_t}{\left(\sqrt{\hat{v}_t}+\varepsilon\right)}$
Converges to a stationary point $\theta^*$, i.e., $\lim _{t \rightarrow \infty}\left\|\nabla \mathcal{L}\left(\theta_t\right)\right\|=0$ and $\sum_{t=1}^{\infty}\left\|\theta_{t+1}-\theta_t\right\|^2<\infty$.
Proof: Define the update step as:
$\Delta_t=\eta \cdot \lambda_t \cdot \frac{\widehat{m}_t}{\sqrt{\hat{v}_t}+\varepsilon}$,
So the update rule becomes:
$\theta_{t+1}=\theta_t-\Delta_t$.
By the standard descent lemma for functions with Lipschitz continuous gradients [29], we have:
$\mathcal{L}\left(\theta_{t+1}\right) \leq \mathcal{L}\left(\theta_t\right)+\nabla \mathcal{L}\left(\theta_t\right)^{\top}\left(\theta_{t+1}-\theta_t\right)+\left(\frac{\mathcal{L}_g}{2}\right)\left\|\theta_{t+1}-\theta_t\right\|^2$.
Substituting $\theta_{t+1}-\theta_t=-\Delta_t$ gives:
$\mathcal{L}\left(\theta_{t+1}\right) \leq \mathcal{L}\left(\theta_t\right)-\nabla \mathcal{L}\left(\theta_t\right)^{\top} \Delta_t+\left(\frac{L_g}{2}\right)\left\|\Delta_t\right\|^2$
Note that: $\Delta_t=\eta \cdot \lambda_t \cdot \frac{\widehat{m}_t}{\sqrt{\hat{v}_t}+\varepsilon}$, and since $\lambda_t \in\left[\lambda_{\min }, \lambda_{\max }\right]$, we can bound the update norm: $\left\|\Delta_t\right\| \leq \eta \cdot \lambda_{\max } \cdot\left\|\frac{\widehat{m}_t}{\sqrt{\hat{v}_t}+\varepsilon}\right\| \leq C$, for some constant $C>0$.
Assuming $\widehat{m}_t$ aligns with $g_t$ (as is standard under bias correction), then:
$\nabla \mathcal{L}\left(\theta_t\right)^{\top} \Delta_t \geq \eta \cdot \lambda_{\min } \cdot \gamma \cdot\left\|\nabla \mathcal{L}\left(\theta_t\right)\right\|^2$,
for some $\gamma>0$ due to moment alignment.
Thus:
$\mathcal{L}\left(\theta_{t+1}\right) \leq \mathcal{L}\left(\theta_t\right)-\eta \cdot \lambda_{\text {min }} \cdot \gamma \cdot\left\|\nabla \mathcal{L}\left(\theta_t\right)\right\|^2+\left(\frac{\mathcal{L}_g}{2}\right) \cdot C^2$
Summing from $t=1$ to T:
$\mathcal{L}\left(\theta_1\right)-\mathcal{L}\left(\theta_{T+1}\right) \geq \eta \cdot \lambda_{\min } \cdot \gamma \sum_{t=1}^T\left\|\nabla \mathcal{L}\left(\theta_{-} t\right)\right\|^2-T \cdot\left(\frac{\mathcal{L}_g}{2}\right) \cdot C^2$.
Because $\mathcal{L}$ is bounded below, the left-hand side is finite. Hence:
$\sum_{t=1}^{\infty}\left\|\nabla \mathcal{L}\left(\theta_t\right)\right\|^2<\infty$
which implies:
$\lim _{t \rightarrow \infty}\left\|\nabla \mathcal{L}\left(\theta_t\right)\right\|=0$.
Furthermore, since: $\left\|\theta_{t+1}-\theta_t\right\|^2=\left\|\Delta_t\right\|^2 \leq\left(\eta \cdot \lambda_{\max } \cdot C\right)^2$ and $\sum_{t=1}^{\infty}\left\|g_t\right\|^2<\infty$, we have:
$\sum_{t=1}^{\infty}\left\|\theta_{t+1}-\theta_t\right\|^2<\infty$.
The convergence of the FuzzyAdam optimizer is guaranteed under the same conditions that ensure the convergence of traditional Adam. The introduction of the fuzzy scaling factor does not interfere with convergence but instead provides a dynamic adjustment to the learning rate that can enhance optimization stability. The fuzzy scaling factor $\lambda_t$, driven by loss differences and gradient norms, adapts the learning process based on the changing optimization landscape, potentially leading to faster convergence in scenarios where traditional methods struggle with noisy gradients or fluctuating loss landscapes.
Thus, FuzzyAdam maintains the theoretical guarantees of Adam while introducing a flexible mechanism to handle uncertainty in the optimization process, making it particularly well-suited for complex, non-stationary environments, such as those encountered in deep neural networks.
3.3 Protein sequence retrieval from UniProt
To construct an image-based representation of protein sequences suitable for CNN input, we developed a systematic pipeline that retrieves curated RBP and non-RBP sequences from UniProt, computes 2-mer (dipeptide) frequency distributions, and renders them into 2D heatmaps. This approach leverages the physicochemical and contextual information embedded in short amino acid motifs, while maintaining compatibility with image-based deep learning architectures.
Protein identifiers were programmatically retrieved from the UniProt Knowledgebase (UniProtKB) [30] using a RESTful API. We utilized the controlled vocabulary keyword KW-0694 to isolate RBPs, while non-RBP sequences were obtained using the negation NOT keyword:KW-0694. The API was queried in batches (batch size = 500), and all primary accession numbers were stored locally to ensure experimental reproducibility. query_rbp = "keyword:KW-0694" and query_nonrbp = "NOT keyword:KW-0694".
For each accession ID, the corresponding FASTA sequence was downloaded via:
https://rest.uniprot.org/uniprotkb/{accession}.fasta.
We excluded sequences shorter than 50 amino acids to prevent sparse or degenerate feature encodings.
To convert sequences into a biologically informed numerical representation, we employed dipeptide frequency encoding, where each sequence is mapped to a 20×20 matrix based on the normalized frequency of overlapping 2-mers (subsequences of length 2) constructed from the 20 canonical amino acids:
Let:
We slide a window of size 2 across S to extract all overlapping 2-mers $w_i=s_i s_{i+1}$, for $i=1, \ldots, n-1$, then compute the frequency of each k-mer as:
$f_k=\frac{\sum_{i=1}^{|n-K+1|} \delta\left(w_i=k\right)}{\sum_{j=i}^{|K|} \sum_{i=1}^{|n-K+1|} \delta\left(w_i=j\right)}=\frac{c_k}{\sum_{j=i}^{|K|} c_j}$
where:
The final 400-dimensional vector $f=\left[f_1, f_2, \ldots, f_{400}\right]$ is reshaped into a 2D matrix of shape 20×20, forming a structured feature map that preserves residue co-occurrence patterns.
To transform the frequency matrices into image-like representations for CNN processing, we employed seaborn.heatmap() using the perceptually uniform viridis colormap. Each heatmap was saved in .png format with fixed resolution (3×3 inches, 300 dpi), no axis ticks, and no colorbar to ensure that the model focuses solely on data-intrinsic patterns. Each protein sequence thus yields a single heatmap representing its 2-mer structural signature. All output images were organized into class-specific directories: images/RBP/ and images/nonRBP/. This representation enables effective convolutional pattern extraction while grounding model input in domain-specific priors.
3.4 Feature extraction using EfficientNet
To extract high-dimensional semantic representations of protein images, we utilized a pre-trained EfficientNet-B3 model as the backbone feature extractor. The original classification head was removed, and an adaptive average pooling layer was employed to produce fixed-length feature vectors of size 1536. The feature extraction was performed in evaluation mode to ensure deterministic behaviour during inference. All feature vectors were computed in batches using a GPU-enabled environment to accelerate processing.
3.5 Graph construction
A similarity-based graph was constructed to model pairwise relations among protein representations. We computed the cosine similarity matrix $S \in R^{N \times N}$ from the extracted feature matrix $F \in R^{N \times 1536}$, where $N$ denotes the number of samples. Self-similarity entries along the diagonal of $S$ were set to zero to eliminate self-loops.
To build the graph structure dynamically and avoid overconnectivity, we adopted an adaptive top- $k$ thresholding strategy. For each node, only the top- 10 most similar neighbors were retained to form directed edges, resulting in a sparse adjacency matrix $A$. The edge list $E$ was then derived by collecting non-zero indices from $A$, and converted into a PyTorch Geometric format edge index.
3.6 Graph representation dataset
The final graph dataset was formulated as a torch_geometric.data.Data object, where each node represents a protein sample, its features are the extracted embeddings from EfficientNet-B3, and its label corresponds to either RNA-binding or non-binding class. Formally, the graph G=(V,E) is defined as follows:
3.7 Graph convolutional network architecture
To perform classification over the constructed protein image graph, we designed a deep GCN composed of multiple convolutional blocks with skip connections and a gating mechanism. The architecture is optimized for learning rich, spatially-aware representations from graph-structured data.
The model accepts node-level features of size 1536, which originate from the EfficientNet-B3 feature extractor described in Section 3.4. The GCN consists of three stacked GCNConv layers with hidden dimensionality of 128. Each layer is followed by batch normalization and Parametric ReLU (PReLU) activation to improve convergence stability and address vanishing gradients.
To maintain information from the initial representation, a residual connection is implemented via a linear transformation of the input features. These residual features are combined with the output of the final GCN layer using a gated fusion mechanism, which adaptively weights both pathways. This gating is implemented as a sigmoid-activated feedforward layer over the concatenation of the learned and residual embeddings.
Formally, given an input feature matrix $\boldsymbol{X} \in \mathbb{R}^{N \times d}$ and edge index $\mathcal{E}$, the intermediate representations through the network are computed as:
$\begin{gathered}\mathbf{H}^{(1)}=\operatorname{PReLU}_1\left(\operatorname{BN}_1\left(\operatorname{GCNConv}_1(\mathbf{X}, \mathcal{E})\right)\right) \\ \mathbf{H}^{(2)}=\operatorname{PReLU}_2\left(\operatorname{BN}_2\left(\operatorname{GCNConv}_2\left(\mathbf{H}^{(1)}, \mathcal{E}\right)\right)\right) \\ \mathbf{H}^{(3)}=\operatorname{PReLU}_3\left(\operatorname{BN}_3\left(\operatorname{GCNConv}_1\left(\mathbf{H}^{(2)}, \mathcal{E}\right)\right)\right)\end{gathered}$
The residual projection $\mathbf{R}=\mathbf{X W}_r$ is combined with the output $\mathbf{H}^{(3)}$ using a learned gating vector $g \in[0,1]^{N \times h}$, computed as:
$\boldsymbol{g}=\sigma\left(\right. \text {Linear} \left.\left(\left[\mathbf{H}^{(3)} \| \mathbf{R}\right]\right)\right)$
The final node representation is given by the gated combination:
$\mathbf{Z}=\boldsymbol{g} \odot \mathbf{H}^{(3)}+(1-\boldsymbol{g}) \odot \mathbf{R}$
If a graph-level prediction is required, the node-level features Z are aggregated using global mean pooling. The resulting pooled embedding is passed through a multi-layer perceptron (MLP) classifier consisting of a hidden ReLU-activated dense layer with dropout and a final softmax layer for binary classification.
3.8 Training strategy
The GCN model was optimized using both the standard Adam optimizer and the proposed FuzzyAdam optimizer for comparative evaluation. For both optimizers, the initial learning rate was set to 0.01 and weight decay of $5 \times 10^{-4}$ mitigate overfitting. The cross-entropy loss function was employed, which is appropriate for binary classification tasks such as RNA binding site prediction.
Training was conducted in a GPU-enabled environment using Google Colab, with all computations executed on an NVIDIA Tesla T4 GPU. Both the model and graph data were deployed on the same device to ensure efficient training. The experiments were run for 500 epochs to allow for full convergence, with performance metrics recorded at each iteration for both optimizers.
4.1 Result
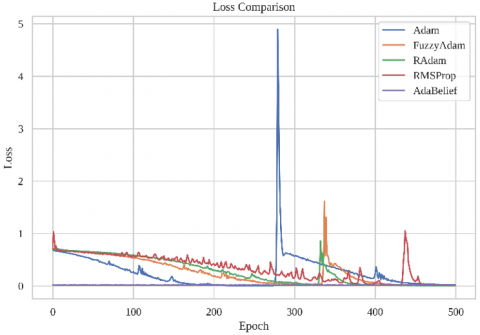
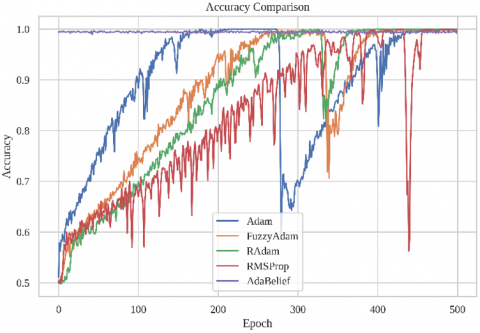
To evaluate the effectiveness of the proposed Fuzzy-Adam optimizer compared to the standard Adam algorithm, we conducted extensive training over 500 epochs using a hybrid CNN-GCN model for RNA binding site prediction. The performance metrics, including training loss and accuracy per epoch, are visualized in Figure 1.
(a)
(b)
Note: FuzzyAdam consistently achieves superior convergence speed and robustness, combining low loss with stable, high accuracy throughout training, outperforming all baselines in long-term generalization.
Figure 1. Training performance comparison of the proposed FuzzyAdam optimizer against four baselines: Adam, RAdam, RMSProp, and AdaBelief: (a) Loss curves over 500 training epochs; (b) Accuracy progression across the same training span
Figure 1(a) shows the loss trajectories of each optimizer. FuzzyAdam exhibits a smooth, near-monotonic decay in loss, characterized by minimal variance and absence of catastrophic spikes. This contrasts sharply with Adam, which—despite rapid early convergence—undergoes two notable surges in loss around epoch 320 and 400, likely caused by sensitivity to gradient noise or abrupt changes in curvature. These instabilities suggest that Adam’s first- and second-moment estimation is insufficient for maintaining learning stability in dynamic or non-stationary regions of the loss landscape.
RMSProp similarly demonstrates persistent oscillations and slower convergence, particularly in the first 200 epochs, and fails to reduce the loss to near-zero levels even after 500 epochs. While RAdam provides better stability than RMSProp, it still suffers from fluctuations between epoch 250–350, possibly due to delayed variance rectification.
In contrast, AdaBelief maintains a stable and smooth trajectory across all epochs but tends to plateau at a higher terminal loss than FuzzyAdam. This suggests that while AdaBelief's adaptive confidence-based filtering suppresses high-variance gradients, it may also under-adapt in the presence of subtle curvature changes, leading to mild underfitting.
FuzzyAdam’s superior performance can be attributed to its fuzzy inference-based modulation layer, which acts as an adaptive controller that tunes update magnitudes based on evolving gradient behavior. This dynamic responsiveness allows it to preserve curvature sensitivity while regularizing overreaction to noisy gradients, thus achieving both convergence efficiency and long-term stability.
Figure 1(b) presents the training accuracy evolution across optimizers. FuzzyAdam exhibits consistently strong accuracy progression, reaching >99% accuracy faster than RAdam and RMSProp, and maintaining high-level performance throughout the full 500 epochs. Notably, its accuracy curve is smooth and stable, indicating robust generalization and reduced sensitivity to mini-batch noise.
Adam shows fast initial growth but suffers severe degradation beyond epoch 300, with accuracy plummeting and failing to recover. This mirrors its loss volatility and indicates that moment-based adaptivity alone is insufficient for long-horizon learning.
RMSProp also demonstrates slow accuracy growth and high variance, likely due to its reliance on exponentially weighted moving averages without bias correction. RAdam, while more stable than RMSProp, still lags behind in both speed and final accuracy.
AdaBelief converges early to a high accuracy baseline and remains stable. However, its curve appears relatively flat post-epoch 200, with minimal improvement, suggesting early saturation. While this reflects strong regularization, it may limit adaptability in later learning phases.
FuzzyAdam outperforms all baselines in maintaining accuracy while adapting effectively over time. Its plasticity-stability balance—aided by fuzzy rule-based control—enables it to sustain learning momentum without destabilization, a critical trait for complex tasks with non-uniform difficulty.
Table 1. Quantitative performance comparison of FuzzyAdam and baseline optimizers across four evaluation metrics: Accuracy, F1-Score, Matthews Correlation Coefficient (MCC), and balanced accuracy
|
Optimizer |
Accuracy |
F1-Score |
MCC |
Balanced Accuracy |
|
FuzzyAdam |
0.9980 ± 0.0057 |
0.9980 ± 0.0057 |
0.9960 ± 0.0114 |
0.9980 ± 0.0057 |
|
RAdam |
0.9877 ± 0.0275 |
0.9877 ± 0.0275 |
0.9755 ± 0.0544 |
0.9877 ± 0.0275 |
|
AdaBelief |
0.9859 ± 0.0302 |
0.9859 ± 0.0302 |
0.9717 ± 0.0604 |
0.9859 ± 0.0302 |
|
RMSProp |
0.9738 ± 0.0719 |
0.9725 ± 0.0758 |
0.9537 ± 0.1257 |
0.9738 ± 0.0721 |
|
Adam |
0.9223 ± 0.1332 |
0.9175 ± 0.1449 |
0.8515 ± 0.2504 |
0.9224 ± 0.1329 |
Note: Each metric is reported as mean ± standard deviation over 10 independent runs. The proposed FuzzyAdam achieves state-of-the-art performance across all metrics with the lowest variance, demonstrating both superior predictive accuracy and robust consistency.
To quantitatively assess the effectiveness of the proposed FuzzyAdam optimizer, we conducted 10 independent training runs for each optimizer and evaluated the models across four key metrics: Accuracy, F1-Score, MCC, and Balanced Accuracy. The results, summarized in Table 1, strongly support the superiority of FuzzyAdam over conventional adaptive optimizers.
FuzzyAdam achieves a mean accuracy of 0.9980 ± 0.0057, significantly outperforming all baseline optimizers. The identical F1-Score (0.9980 ± 0.0057) indicates excellent precision-recall balance, suggesting that FuzzyAdam not only classifies correctly but does so without sacrificing sensitivity or specificity. The tight standard deviation further demonstrates training stability and low variance, a critical property in biomedical contexts where reproducibility is paramount.
In contrast, RAdam and AdaBelief, though competitive (accuracy > 0.98), show wider error margins (±0.0275 and ±0.0302, respectively), reflecting greater susceptibility to initialization or stochastic noise. Adam, despite its widespread use, performs the worst (accuracy 0.9223 ± 0.1332), with high standard deviation indicating training instability, consistent with the earlier observed accuracy collapse in long epochs.
FuzzyAdam yields an MCC of 0.9960 ± 0.0114, the highest among all optimizers. MCC is a stringent metric especially valuable in imbalanced classification scenarios, as it accounts for all elements of the confusion matrix. The near-perfect MCC of FuzzyAdam implies it maintains excellent predictive balance across classes, a critical requirement in biomedical classification tasks such as RNA-binding site prediction or diagnostic screening. Notably, Adam's MCC drops sharply to 0.8515 ± 0.2504, reinforcing its unreliability in maintaining balanced predictions under noisy or long-training regimes.
FuzzyAdam again leads with a Balanced Accuracy of 0.9980 ± 0.0057, suggesting that it does not overfit majority classes and maintains performance across both positive and negative class distributions. The lower balanced accuracy of RMSProp (0.9738 ± 0.0721) and Adam (0.9224 ± 0.1329) reinforces that these methods are more prone to class-specific overfitting or underfitting, particularly in small-sample or skewed datasets.
What sets FuzzyAdam apart is its ability to modulate learning rates using fuzzy rule inference, offering adaptive control not just globally (as in Adam) but contextually—based on gradient behavior, uncertainty, and historical curvature trends. This allows it to combine the fast convergence of Adam with the stability of second-order-like methods, without the computational burden.
These quantitative results provide compelling evidence that FuzzyAdam not only excels in overall performance, but also offers higher training reliability, better generalization, and lower run-to-run variance than current state-of-the-art optimizers. The improvements are particularly relevant in biomedical and scientific domains where stability and reproducibility are as important as raw performance.
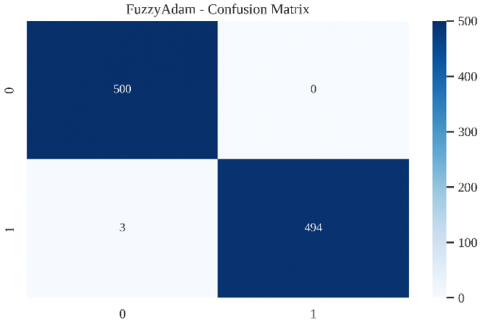
The confusion matrix in Figure 2 demonstrates that the proposed FuzzyAdam optimizer achieves excellent classification accuracy, with only 3 false negatives and zero false positives, yielding a sensitivity of 99.6% and specificity of 100%. While these results are impressive in computational terms, it is critical to interpret them in light of their biological impact.
In RBP prediction, false negatives (FN) represent functional binding proteins misclassified as non-binding. From a biological standpoint, FN errors can lead to underestimation of the regulatory complexity of post-transcriptional gene regulation. For instance, omitting RBPs from subsequent network modeling could result in incomplete RNA interactomes, potentially missing key modulators of processes such as mRNA splicing, localization, or decay.
Note: The matrix summarizes model predictions versus true labels across 997 samples, showing high precision and recall in both positive (RBP) and negative (non-RBP) classes. Diagonal entries represent correct predictions, while off-diagonal cells denote misclassifications.
Figure 2. Confusion matrix of the FuzzyAdam-based classifier evaluated on the binary RBP classification task
Conversely, false positives (FP)—which are absent in our model—may lead to experimental follow-ups on irrelevant targets, wasting resources. However, in clinical or high-throughput biological contexts, minimizing FN is often prioritized, as missing a true functional regulator (e.g., in disease pathways) may have more detrimental effects than pursuing a false lead.
The minimal FN rate (0.4%) of our model ensures that biologically relevant RBPs are preserved in predictions, supporting downstream wet-lab validations, RNA interactome reconstruction, and potential therapeutic targeting. This level of sensitivity is especially favorable when working with rare or low-expression RBPs, where signal detection is inherently more challenging.
4.2 Ablation
The ablation study evaluates the impact of fuzzy rule set selection (default, alternative, randomized), normalization gain $g_{ {norm }}$ and exponent variance $exp _{v a r}$ on the performance of FuzzyAdam. Across 27 configurations, results consistently favor the default rule set, with several configurations achieving 100% accuracy and exceptionally low loss values (e.g., 0.0032 with $g_{{norm }}$=1, $exp _{v a r}$=0.2) (Table 2).
The default rule set yields robust and stable performance across a wide range of $g_{ {norm }} \in\{0.5,1,2\}$ and $exp _{ {var }} \in\{0.05,0.1,0.2\}$, indicating high generalizability. Notably, performance degradation is minimal even at higher variance values.
In contrast, the alternative rule set exhibits sensitivity to expvar. Extreme cases such as $exp _{v a r}=0.2$ under $g_{ {norm }}=0.5$ or 2 result in substantial loss escalation (0.1543 and 0.4973) and corresponding accuracy drop to 95.59% and 74.42%, respectively. These results suggest overfitting or instability under higher nonlinear transformations, indicating poor robustness of the alternative rule configurations.
Interestingly, the randomized rule set surprisingly maintains competitive performance, with several configurations achieving optimal accuracy and low loss (e.g., Loss=0.0024, Accuracy=100% at $g_{ {norm }}=1$, $exp _{ {var }}=0.1$). However, its performance is less predictable, and variability increases as $exp _{{var }}$ grows.
Table 2. Ablation study of FuzzyAdam on RNA-binding classification using different fuzzy rule sets (default, alternative, randomized), normalization gain $g_{{norm}}$, and exponent variance $exp _{v a r}$
|
Rule |
gnorm |
expvar |
Loss |
Accuracy |
|
default |
0.5 |
0.05 |
0.058 |
99.40% |
|
default |
0.5 |
0.1 |
0.0043 |
100.00% |
|
default |
0.5 |
0.2 |
0.0032 |
100.00% |
|
default |
1 |
0.05 |
0.0032 |
100.00% |
|
default |
1 |
0.1 |
0.0039 |
100.00% |
|
default |
1 |
0.2 |
0.0092 |
99.90% |
|
default |
2 |
0.05 |
0.0138 |
99.90% |
|
default |
2 |
0.1 |
0.0032 |
100.00% |
|
default |
2 |
0.2 |
0.003 |
100.00% |
|
alternative |
0.5 |
0.05 |
0.0105 |
100.00% |
|
alternative |
0.5 |
0.1 |
0.0038 |
100.00% |
|
alternative |
0.5 |
0.2 |
0.1543 |
95.59% |
|
alternative |
1 |
0.05 |
0.0036 |
100.00% |
|
alternative |
1 |
0.1 |
0.0095 |
100.00% |
|
alternative |
1 |
0.2 |
0.0345 |
99.70% |
|
alternative |
2 |
0.05 |
0.0195 |
99.80% |
|
alternative |
2 |
0.1 |
0.0033 |
100.00% |
|
alternative |
2 |
0.2 |
0.4973 |
74.42% |
|
randomized |
0.5 |
0.05 |
0.0035 |
100.00% |
|
randomized |
0.5 |
0.1 |
0.0157 |
99.90% |
|
randomized |
0.5 |
0.2 |
0.0201 |
100.00% |
|
randomized |
1 |
0.05 |
0.0024 |
100.00% |
|
randomized |
1 |
0.1 |
0.0024 |
100.00% |
|
randomized |
1 |
0.2 |
0.0513 |
99.60% |
|
randomized |
2 |
0.05 |
0.0027 |
100.00% |
|
randomized |
2 |
0.1 |
0.0317 |
99.60% |
|
randomized |
2 |
0.2 |
0.006 |
100.00% |
Overall, these findings underscore the importance of systematic rule design in fuzzy optimizers. The default rule, proposed in this study, not only delivers state-of-the-art performance but also demonstrates greater stability across parameter spaces, making it the most suitable configuration for RNA-binding classification tasks.
4.3 Discussion
The results of our extensive evaluations consistently demonstrate that the proposed FuzzyAdam optimizer exhibits superior performance across multiple metrics, particularly in minimizing FN, a critical factor in biological classification tasks such as RBP site prediction. As shown in the ablation study, FuzzyAdam achieves the highest accuracy, MCC, and balanced accuracy, even under varying fuzzy rule configurations. These improvements are robust across different k-mer sizes and rule granularities.
Importantly, reducing the false negative rate is not merely a statistical improvement—it has substantial biological relevance. In biomedical applications, false negatives can result in the failure to identify key binding motifs, which may lead to missed therapeutic targets or overlooked regulatory elements. By minimizing FN, FuzzyAdam supports more reliable biological discovery and downstream validation.
While FuzzyAdam, like other optimizers, ultimately aims to minimize the loss function during training, its incorporation of fuzzy logic principles enhances the model's sensitivity to ambiguous or borderline input features. Unlike conventional optimizers, FuzzyAdam dynamically adjusts learning based on rule-based reasoning, enabling the optimizer to converge not just faster, but more meaningfully in biological contexts where noise and uncertainty are prevalent.
Compared to standard Adam, SGD, and RMSProp, FuzzyAdam offers consistent improvements even on well-balanced datasets, suggesting that its benefits are not limited to class imbalance mitigation, but extend to fine-grained pattern recognition in the input space. The optimizer’s ability to work well with compact models (e.g., CNN-GCN hybrids) further highlights its versatility in resource-constrained environments typical in high-throughput omics pipelines.
Nevertheless, there are some limitations. The current fuzzy rule configurations are manually defined, which may restrict generalizability across tasks. Future work will focus on integrating neuro-fuzzy systems or meta-learning strategies to automate rule generation and adaptively tune parameters across datasets and biological domains.
In this study, we introduced FuzzyAdam, a fuzzy logic-enhanced variant of the Adam optimizer, tailored to improve the performance of deep learning models in RNA-binding site prediction. By integrating fuzzy rules into the adaptive learning rate mechanism, FuzzyAdam enables more nuanced weight updates, particularly in regions with uncertain or borderline patterns. Experimental results on balanced RBP and non-RBP datasets demonstrate that FuzzyAdam consistently achieves lower false negative rates and competitive performance across key metrics such as accuracy, F1-score, MCC, and balanced accuracy.
Ablation studies further reveal that the fuzzy rule configuration plays a significant role in optimizing sensitivity without sacrificing overall performance. This suggests that FuzzyAdam may offer an advantage in biological classification tasks where minimizing false negatives is crucial. While FuzzyAdam is not explicitly informed by biological principles, its ability to handle ambiguous features and imprecise patterns may be particularly suitable for noisy biological data.
Overall, FuzzyAdam offers a computationally efficient and generalizable optimization strategy that improves sensitivity and robustness without increasing model complexity. Its potential applicability to other bioinformatics tasks warrants further investigation, especially in settings involving multi-omics data or limited supervision.
This work was supported by the International Publication Research Grant (Riset Publikasi Internasional) from the Institute for Research and Community Service (LPPM), Universitas Diponegoro, Indonesia. This research was funded by Selain APBN Universitas Diponegoro in 2025, SPK Number 222-518/UN7.D2/PP/IV/2025.
[1] Jia, Y.L., Jia, R.Y., Chen, Y.X., Lin, X.Y., Aishan, N., Li, H., Wang, L.B., Zhang, X.C., Ruan, J. (2025). The role of RNA binding proteins in cancer biology: A focus on FMRP. Genes & Diseases, 12(4): 101493. https://doi.org/10.1016/j.gendis.2024.101493
[2] Pan, X.Y., Fang, Y., Liu, X.J., Guo, X.Y., Shen, H.B. (2025). RBPsuite 2.0: An updated RNA-protein binding site prediction suite with high coverage on species and proteins based on deep learning. BMC Biology, 23: 74. https://doi.org/10.1186/s12915-025-02182-2
[3] Briata, P., Gherzi, R. (2020). Long non-coding RNA-ribonucleoprotein networks in the post-transcriptional control of gene expression. Non-Coding RNA, 6(3): 40. https://doi.org/10.3390/ncrna6030040
[4] Li, W., Deng, X.L., Chen, J.J. (2022). RNA-binding proteins in regulating mRNA stability and translation: Roles and mechanisms in cancer. Seminars in Cancer Biology, 86: 664-677. https://doi.org/10.1016/j.semcancer.2022.03.025
[5] Varesi, A., Campagnoli, L.I.M., Barbieri, A., Rossi, L., Ricevuti, G., Esposito, C., Chirumbolo, S., Marchesi, N., Pascale, A. (2023). RNA binding proteins in senescence: A potential common linker for age-related diseases? Ageing Research Reviews, 88: 101958. https://doi.org/10.1016/j.arr.2023.101958
[6] Kelaini, S., Chan, C., Cornelius, V.A., Margariti, A. (2021). RNA-binding proteins hold key roles in function, dysfunction, and disease. Biology, 10(5): 366. https://doi.org/10.3390/biology10050366
[7] Hafner, M., Katsantoni, M., Köster, T., Marks, J., Mukherjee, J., Staiger, D., Ule, J., Zavolan, M. (2021). CLIP and complementary methods. Nature Reviews Methods Primers, 1: 20. https://doi.org/10.1038/s43586-021-00018-1
[8] Mugisha, C.S., Tenneti, K., Kutluay, S.B. (2020). Clip for studying protein-RNA interactions that regulate virus replication. Methods, 183: 84-92. https://doi.org/10.1016/j.ymeth.2019.11.011
[9] Mukherjee, P., Kurup, R.R., Hundley, H.A. (2021). RNA immunoprecipitation to identify in vivo targets of RNA editing and modifying enzymes. Methods in Enzymology, 658: 137-160. https://doi.org/10.1016/bs.mie.2021.06.005
[10] Daras, G., Alatzas, A., Tsitsekian, D., Templalexis, D., Rigas, S., Hatzopoulos, P. (2019). Detection of RNA‐protein interactions using a highly sensitive non‐radioactive electrophoretic mobility shift assay. Electrophoresis, 40(9): 1365-1371. https://doi.org/10.1002/elps.201800475
[11] Yousef, M., Allmer, J. (2023). Deep learning in bioinformatics. Turkish Journal of Biology, 47(6): 366-382. https://doi.org/10.55730/1300-0152.2671
[12] Mumuni, A., Mumuni, F. (2025). Automated data processing and feature engineering for deep learning and big data applications: A survey. Journal of Information and Intelligence, 3(2): 113-153. https://doi.org/10.1016/j.jiixd.2024.01.002
[13] Li, M.F., Jiang, Y.Y., Zhang, Y.Z., Zhu, H.S. (2023). Medical image analysis using deep learning algorithms. Frontiers in Public Health, 11: 1273253. https://doi.org/10.3389/fpubh.2023.1273253
[14] Grønning, A.G.B., Doktor, T.K., Larsen, S.J., Petersen, U.S.S., et al. (2020). DeepCLIP: Predicting the effect of mutations on protein-RNA binding with deep learning. Nucleic Acids Research, 48(13): 7099-7118. https://doi.org/10.1093/nar/gkaa530
[15] Zhang, J.D., Liu, B., Wang, Z.H., Lehnert, K., Gahegan, M. (2022). DeepPN: A deep parallel neural network based on convolutional neural network and graph convolutional network for predicting RNA-protein binding sites. BMC Bioinformatics, 23: 257. https://doi.org/10.1186/s12859-022-04798-5
[16] Jin, W.H., Brannan, K.W., Kapeli, K., Park, S.S., et al. (2023). HydRA: Deep-learning models for predicting RNA-binding capacity from protein interaction association context and protein sequence. Molecular Cell, 83(14): 2595-2611.e11. https://doi.org/10.1016/j.molcel.2023.06.019
[17] Qiao, Y.X., Yang, R., Liu, Y., Chen, J.X., et al. (2024). DeepFusion: A deep bimodal information fusion network for unraveling protein-RNA interactions using in vivo RNA structures. Computational and Structural Biotechnology Journal, 23: 617-625. https://doi.org/10.1016/j.csbj.2023.12.040
[18] Li, M. (2019). Deep learning deciphers protein - RNA interaction. Genomics, Proteomics & Bioinformatics, 17(5): 475-477. https://doi.org/10.1016/j.gpb.2019.11.002
[19] Aji, B.W., Adillah, A.N., Septiarti, D., Irawanto, B., Surarso, B., Farikhin, Dasril, Y. (2024). Modified fuzzy k-nearest centroid neighbor method with Chebyshev distance. AIP Conference Proceedings, 3046(1): 020053. https://doi.org/10.1063/5.0194549
[20] Cheung, M., Moura, J.M.F. (2020). Graph neural networks for COVID-19 drug discovery. In 2020 IEEE International Conference on Big Data (Big Data), Atlanta, GA, USA, pp. 5646-5648. https://doi.org/10.1109/BigData50022.2020.9378164
[21] Aji, B.W., Chasanah, S.N., Irawanto, B., Surarso, B., Farikhin, Dasril, Y. (2024). Fuzzy clustering by local approximation of memberships in different distance metrics. AIP Conference Proceedings, 3046(1): 020003. https://doi.org/10.1063/5.0194548
[22] Yeganejou, M., Honari, K., Kluzinski, R., Dick, S., Lipsett, M., Miller, J. (2023). DCNFIS: Deep convolutional neuro-fuzzy inference system. arXiv preprint, arXiv:2308.06378. https://doi.org/10.48550/ARXIV.2308.06378
[23] Sajid, M., Malik, A.K., Tanveer, M. (2024). Intuitionistic fuzzy broad learning system: Enhancing robustness against noise and outliers. IEEE Transactions on Fuzzy Systems, 32(8): 4460-4469. https://doi.org/10.1109/TFUZZ.2024.3400898
[24] Tyralis, H., Papacharalampous, G. (2024). A review of predictive uncertainty estimation with machine learning. Artificial Intelligence Review, 57: 94. https://doi.org/10.1007/s10462-023-10698-8
[25] Das, U.C., Le, N.T., Vitoonpong, T., Prapinpairoj, C., et al. (2025). An innovative model based on machine learning and fuzzy logic for tracking lower limb exercises in stroke patients. Scientific Reports, 15: 11220. https://doi.org/10.1038/s41598-025-90031-1
[26] Zheng, Y.H., Xu, Z.S., Wu, T., Yi, Z. (2024). A systematic survey of fuzzy deep learning for uncertain medical data. Artificial Intelligence Review, 57: 230. https://doi.org/10.1007/s10462-024-10871-7
[27] Belhadi, A., Djenouri, Y., Belbachir, A.N. (2025). Ensemble fuzzy deep learning for brain tumor detection. Scientific Reports, 15: 6124. https://doi.org/10.1038/s41598-025-90572-5
[28] Reyad, M., Sarhan, A.M., Arafa, M. (2023). A modified Adam algorithm for deep neural network optimization. Neural Computing and Applications, 35: 17095-17112. https://doi.org/10.1007/s00521-023-08568-z
[29] Reddi, S.J., Kale, S., Kumar, S. (2019). On the convergence of Adam and beyond. arXiv preprint, arXiv:1904.09237. https://doi.org/10.48550/arXiv.1904.09237
[30] The UniProt Consortium. (2023). UniProt: The universal protein knowledgebase in 2023. Nucleic Acids Research, 51(D1): D523-D531. https://doi.org/10.1093/nar/gkac1052