Saroj Shambharkar* | K. Vaishali | Rachna Somkunwar | Yogeshri Choudhari | Jyotsna Gawai
© 2022 IIETA. This article is published by IIETA and is licensed under the CC BY 4.0 license (http://creativecommons.org/licenses/by/4.0/).
OPEN ACCESS
In system biology inference from gene regulatory network (GRN) is a challenging task. There exist different computational techniques to analyze the causal relationships between the pair of genes and to understand the significance of causal relationship in gene regulatory network. The DREAM4 insilico network structure and insilico gene expression time series dataset of DREAM challenge dataset is examined. This gene expression dataset of insilico of size 10 is analyzed for inferring causal relationships of the GRN inference. The analysis of dataset showing the gene expression data values are varying with respect to time. The paper focused on the different models of causal inference approach, Genetic Algorithm framework for the GRN inference. In this dataset, values associated with genes are analyzed using the Granger causality test and clustering to analyze the correlation and interaction or causal relationships among genes. The objective behind analysis and inferring causal information in the GRN is to reveal the study on gene activities to achieve more biological insights.
biological network, clustering, casual relationships, GeneNetWeaver, gene regulatory network (GRN), Granger causality
In bioinformatics, there are different biological networks like gene regulatory network, Protein-Protein Interaction (PPI), Biochemical, transcriptional regulation, signal transduction and metabolite networks which are used to describe the biological processes occurring within the cell. The generalized biological network, consist of nodes as genes, proteins, metabolites, enzymes and organisms and edges can be interactions, regulations, reactions, transformation, activations, inhibition etc.
Definition 1.1A Gene regulatory network (GRN) is consisting of nodes and edges. The edges can be regulatory links and vertices can be Transcription Factors (TF’s) or Target Genes (TG’s). A Gene Regulatory Network is a cellular network can be represented using directed or undirected graph. The GRN represented as graph Gi=<G, I>, G comprises of vertices or nodes represent them as genes and I comprises of edges represent them as causal relationship among genes.
Definition 1.2 The transcriptional regulatory network consist of genes, proteins, mRNA, metabolites, etc. and these components are used to construct various models like Protein-Protein Interaction model (PPI), GRN, etc. The Transcription Factor’s control activation of the genes called as transcription rate.
Definition 1.3 Co expression Network is obtained from the GRN by calculating the Co expression between two genes.
There are proximal genes (highly co expressed) and distal genes (low co expressed). The Co expression identifies linear dependence by measuring correlation and non-linear dependence by Mutual Information (MI) between pair of genes. Both correlation and MI can be used to infer the network. It is demonstrated in the example given below is used to estimate the MI is amount of information shared among two genes say X and Y:
MI (XY)=H(gene X) +H(gene Y) - H (gene X and gene Y)
In above estimation, H(X) is the Entropy of gene X, H(Y) is the Entropy gene Y and H (XY) is the Joint distribution of both genes. The Co expression network consists of direct and indirect interaction and it is found that direct interaction has high Co expression as compared to indirect interaction.
The paper is organized in different sections as given below. The section II describes the work carried out by different researchers on inference techniques, approaches, models and algorithms implemented for analyzing the GRN and inferred network from GRN using various approaches. In section III the different types of causal relationships types of the GRN are described with example. In section IV the classification of different causal approaches and models coming under each approach is discussed. In section V, the Gene Regulatory Inference using Boolean Network is discussed. The section VI the Gene Regulatory Inference using Genetic Algorithm (GA) along with operator and fitness estimation of target gene in its subsections is discussed. The section VII discusses the analysis of DREAM challenge dataset using Granger causality test and correlation matrix. In section VIII the conclusion of GRN inference on gene expression data is discussed and future work is mentioned.
The method Genetic Algorithm Based Network Inference (GABNI), the Boolean function implemented for interaction is used for searching regulatory genes in large search space. This work implemented their method on artificial, time series gene expression and real gene expression dataset. The structural and dynamic accuracy obtained by GABNI approach is outperformed compared with MIBNI, ARACNE, TIGRESS and GENIE3 methods. The inferred Boolean network obtained from the time series Gene expression dataset [1, 2]. A model called a GripDL constructed the GRN for Drosophilla eye development by taking input as Drosophilla embryonic gene expression images [3].
From the literature survey, it is identified the different challenges encountered with GRN were mentioned and the solution of them. Maximum research work for the GRN inference is carried out on network structure from DREAM challenge and time-course data based on expression values of different set of genes. The research work is carried out on causal inference approach based on cancer disease and other interactions inference happened in cell, also most of the research work carried out on microarray experimental gene expression dataset and on RNA-seq data. The different categories of computational models for the GRNs inference are used and their performance also compared. They used Continuous model, Logical model, Probabilistic model, Interaction Information Model, Algebraic model, Statistical and Stochastic Network, and Hybrid model. The computational methods are less time consuming and cost efficient. From the literature, the different Discretization methods are discussed in Table I used in preprocessing of Gene Expression dataset. The approach towards Discretization is used by many inference techniques of the GRN is given in Table 1.
Table 1. Types of decentralization methods
|
Discretization method |
Approach |
|
Equal Frequency Discretization (EFD) |
Partition all gene expression values of every gene into equal size partitions. |
|
Equal Width Discretization (EWD) |
Partition all gene expression values of every gene into n bins of equal size. |
|
Global Frequency Discretization (GFD) |
Partition all gene expression values of every gene in dataset into equal size partitions. |
|
RowkMeans |
Partition values of every gene into k clusters. |
|
ColkMeans |
Partition all gene expression values based on condition into k clusters. |
|
BikMeans |
Partition the gene expression values based on both RowkMeans and ColkMeans. |
The study of various Discretization methods is useful in preprocessing of high dimensional gene expression dataset. The advantage of preprocessing is to improve the data quality and helpful in better data analysis as compared to the data without preprocessing.
For GRN inference, study of interactions and causal relationships between the pair of nodes is important for the biological insight which is a challenging task. The causal relationship inference gives the information about the functioning of genes within a cell. In GRN, the interactions or edges can be directed or indirect, this gives us causal information which explains the gene activities within the cell. If there exist an undirected edge between a pair of genes it indicates they are influenced by each other, and if there is direct edge <G0, G1> gene G0 to gene G1, it represents the gene G1 is influenced by gene G0. The direct causal relationship from G0 to G1 also represents that G0 is a parent gene and G1 is an offspring.
In directed GRN, an edge represents a change in the offspring gene following perturbation of the parent gene.
Depending upon the type of direct or indirect interactions or causal relationships between the genes within cell is viewed through GRN. There may exist following 6 types of mapping or causal relationships:
The causal interactions or mappings are explained with reference to the Gene Regulatory Network (GRN) represented in the form of graph. The graph shown in Figure 1 is insilico Network structure of DREAM4 obtained from Gene NetWeaver. The network structure, GRN represented as graph consists of set of genes from G1 to G10 and 15 interactions or causal relationships.
The different types of causal relationships observed in the network structure of Figure 1 are explained below: -
The extraction and inferring causal relationship between the pair of genes from the GRN helps in understanding the cause of complex human diseases. The Genes with high out degree value are influenced by maximum number of other genes and cause of a disease. It is helpful in making decision in progression of complex human diseases like cancer, AIDS, neurodegenerative diseases [4].
There are various methods to infer causal relationship in GRN. They are Boolean networks, Bayesian Networks, Dynamic Bayesian Networks, Granger Causality, Transfer Entropy, Interaction Information or Conditional Mutual Information and Causation Entropy.
For causal inference, the DREAM4 insilico, Ecoli and Yeast network structure can be obtained from Gene NetWeaver. The Gene NetWeaver is a Java based platform, it provides methods for both insilico benchmark generation and performance profiling of network inference algorithms. This framework is helpful to obtain the GRNs and sub networks from existing networks, available in Gene NetWeaver.
Figure 1. Graph (G) is representation of the DREAM4 Insilco network structure to demonstrate the causal relationships
The problem of causal inference begins with treatment. The causal inference makes the prediction models most robust. The causal inference helps to evaluate impact of systems. The Causal inference task is divided into 3 parts, first is discovering the causal model from the data, second is identifying the causal effect from known causal effect and third is estimating a causal effect from the data.
Figure 2. Classification of causal inference approach
There are three broad categories of causal Inference approaches to infer the GRN are model based approach, data driven approach and multi-network approach. The detail classification is elaborated in Figure 2. The data driven and model-based methods are widely used because of its simplicity, accuracy and computational efficiency [5].
A. Model Based Approach
The model-based approach is based on hypothesis and parameters. For the GRN inference using this type of approach, the model is fitted using experimental data. The probabilistic and dynamic models are based on model based approach. In probabilistic model, the fluctuations in gene expression level are considered to model the GRN [6]. The dynamic model can make use of time series gene expression data, insilico network for inference.
B. Data Driven Approach
In data driven based model approach, the interaction dependency between the pair of genes in GRN is estimated. The data driven approach uses two types of scores, correlation score and Information Theory score for estimating the link dependency. The correlation score for simple relationships and Information Theory score for complex relationships. The output of data driven approach is dependent on scoring function.
The probabilistic uses two types of models, Bayesian Network and Gaussian Graphical models.
The Bayesian networks are associated with two issues, first is there can be many potential parent sets and second is network inferred using Bayesian network approach it should not contain any cycles. The second issue is coming under NP-complete problem.
The Gaussian graphical model is a probabilistic model. To model the GRN using Gaussian graphical model it makes use of log transformed gene expression profiles. Using Gaussian Graphical model, in GRN each node is expressed gene, and interaction is the conditional dependence between the pair of expressed genes [7].
The dynamic Bayesian networks, ordinal differential equations, Boolean networks and neural networks are dynamic models. The Dynamic networks are variants of Bayesian Network models. They describe the time dependent relationships between the nodes.
Figure 3. Dynamic Bayesian network
The dynamic network from simple sub network is represented in above (Figure 3) the given structure is represented in terms of two instants of time t (before) and t+1 (after). One gene with different versions shown in Figure 3. In Figure 1 the one to one causal relationship is indicating the gene G2 is getting influenced by G2 after some time, the reason maybe changes in the cell after some time period due to some environmental condition or because of some other state of genes.
The Ordinal Differential Equation framework comprises of differential equations. The differential equation is expressed in terms of one independent variable and 1 or more of their derivatives with respect to the variable [8].
The Ordinal Differential Equation Model is a deterministic model. Using Ordinal Differential Equation, the nodes represent genes interaction as causal interactions in GRN, the interactions are not representing statistical independence [9, 10].
The Boolean network is deterministic and dynamic network. Using Boolean Network approach, the GRN model is using genes and their interactions. The genes are represented as 0 (unexpressed) or 1(expressed) and interactions represent Boolean functions. The MIBNI it is specified for use in small search space lack into obtain optimal solution and GABNI gives optimal solution for huge problem space Boolean network inference from the given time-course gene expression dataset [1]. Another limitation with MIBNI algorithm was the Boolean function is limited to conjunction and disjunction operation to represent interactions.
C. Multi-network model approach
The multi-network model-based inference approach uses different data sources. The data sources can be gene expression data, TF binding site motifs, or Chromatin Immuno-Precipitation Data [5].
Out of all mentioned causal inference models in Figure 1, discussed the inferring Boolean network model using genetic algorithm. The definition of Boolean Network is modified to relate with time series gene expression dataset. The data values of given challenge dataset of gene expression having 10 attributes G1 to G10 associated with gene expression values at different time instant. In Boolean network interaction is represented by Boolean function f. For all 10 genes, the number of combinations possible for Boolean model will be calculated as 2 2*10. A node ith gene, Gi at time t is denoted as Gi(t) and at t+1 as Gi(t+1). The Boolean function for Gi(t+1) is function of n regulatory genes at some instant t. The function defined as fi(Gi1(t), Gi2(t), Gi3(t), Gi4(t), Gi5(t), Gi6(t),………, Gin(t)) [1].
For the given GRN, depending on type of edge or relationship, number of interactions, the Boolean function for each vertex (gene) in the network can be defined.
The Boolean network model can be used to infer the GRN. The inferred network can be analyzed based on dynamic and structural accuracy. The dynamic accuracy is estimated from consistency accuracy using Eq. (1).
Accuracy=[∑ni=1 C(Gi,Gi’)]/N (1)
where, N is total genes in dataset and C (Gi,Gi’)is dynamic consistency is similarity between the Boolean trajectories of the observed gene expression G(t) and the estimated gene expression G’(t).
The true network (GRN) has shown in Figure 4 used for inference using Boolean network approach.
When the Boolean Network Learning model applied on true network presented in Figure 5, the AUC, Area under Curve estimated as 0.09523809523809523.
Figure 4. True network considered for GRN inference using Boolean Network model
Figure 5. Boolean network based gene regulatory network
The following steps required using GA approach with reference to the GRN inference.
Step 1: Input the time series dataset and the genes in dataset will be consider for formation of population.
Step 2: Initialization
Initialize the population with a set of chromosomes, each chromosome consisting of n number of genes. The encoding for genes is 1 for regulatory genes and others are set to 0.
Step 3: Selection
From the population two chromosomes as parent 1 (pc1) and parent 2 (pc2) using roulette wheel selection process.
Step 4: Generation of offspring’s
Generate offspring’s O1 and O2 by applying crossover operator on selected parent chromosomes from step1.
Step 5: Mutation
Apply mutation operator on the offspring’s O1 and O2. The resultant offspring’s after mutation is stored as O11 and O22.
Step 6: Update Population
The parent chromosome of step1pc1 and pc2 are replaced with O11 and O22.
Step 7: Repeat steps from 2 through 5 until we get optimal solution.
Step 8: Obtain inferred network
Operators of GA in GRN inference.
The operators used in GRN inference using GA based approach are selection, crossover and mutation.
The Genetic Algorithm started with population of set of regulatory genes. From the population the chromosomes are selected using roulette wheel selection method. The selection process is directly proportional to fitness value and the chromosome with high fitness value is selected from the population. The fitness is directly proportional to the maximum area (high fitness value portion) of the roulette wheel and it will take part in GA.
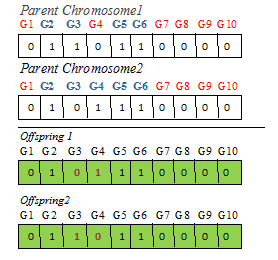
The crossover operator illustrated using step 4 in Figure 6. The genetic Algorithms are used to represent a computational method. It evaluates a set of solution and hypothesis called population. The GA generates best solutions by applying mutation operation from existing population (solutions and hypotheses) [11].
To infer GRN using GA framework. The framework uses crossover and recombination, mutation, and selection genetic operators. The genetic operator crossover is applied on parent chromosomes consisting of 10 genes.
Figure 6. Generation of offspring’s using Crossover operator applied on G1 to G10 (parent chromosomes)
A. Fitness estimation of target gene using regulatory genes
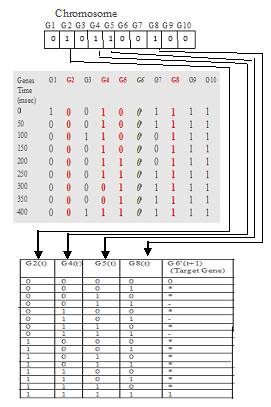
A chromosome is encoded in the form of 0’s and 1’s (Boolean network approach) and the number of bits required to represent the chromosome is equal to total regulatory genes. The bit in representation of regulatory genes is equal to 1 and others as 0 shown in Figure 7. These excluded bits of chromosome do not affect the dynamic consistency. For example, if there are 10 genes (G1 to G10) and regulatory genes considered as G2, G4, G5 and G8. Then the chromosome is represented as:
Figure 7. Representation of chromosome: regulatory genes (bit 0) and target genes (bit 1)
The chromosome encoding for regulatory genes G2, G4, G5, G8 and G6 as target gene, the rules are generated as shown in Figure 8 denoted in column G6’(t+1).
The target gene column G6’ (t+1) indicates 4 different values 0, 1,*,-.The fitness values * and – does not affect dynamic consistency and indicates the bit pattern is not found in binary Gene expression dataset. The binary gene expression dataset is taken from the insilico time series gene expression dataset, where the threshold value taken as 0.5. The values below 0.5 are set to 0 otherwise 1. The ‘– ‘symbol is used when there is tie between 0 and 1 value. And for regulatory genes with binary string 0000 and 1111 the fitness value is 0 and 1 respectively.
Figure 8. Demonstration of rule generation and fitness estimation
The fitness value is dependent on dynamic consistency, number of regulatory genes, k and weight factor γ. The fitness F value of a chromosome is evaluated as:
F=1/ [((1-dynamic consistency).γ)+k]
The fitness value of chromosome is large, for small value of k and (1-dynamic consistency). γ) >> k.
As the steps mentioned for GA, we select any two parent chromosomes from the population of binary time series gene expression dataset [1].
The Gene Net Weaver (GNW) simulator is providing the gene expression dataset. In GNW the network desktop providing the different storage containing the GRN’s, from there we have selected the DREAM_challenges. The DREAM_challenges consist of DREAM3 and DREAM4insilico of network size 10, 100, 50 and these networks are containing the network of Ecoli and Yeast. After deciding the size required for analysis the time series dataset equivalent to it is generated by specifying the time points, we wish for analysis. The DREAM is a Dialogue on Reverse Engineering Assessment and Methods. It provides different datasets. The DREAM challenge has provided network of 10, 50 and 100 nodes. The time series gene expression dataset of insilico downloaded from challenge dataset of DREAM 4 consists of 21 time points set and the number of times 21 points repeated depending in the dataset [12].
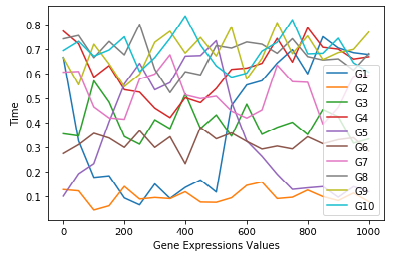
The changes in gene expression values are shown in Figure 9a for first 21 time points for 10 genes G1 through G10.
It has been observed from the dataset, the gene expression values has been changed in every set of 21 time points for G1 to G10. The DREAM4 is a framework also provided other three time-series dataset of Ecoli, Yeast. The DREAM is running many challenges including molecular network inference.
From Figures 9a and 9b the gene expression values are changing with time course within a cell, they are not static.
(a)
(b)
Figure 9. (a) Representation of change in G1 to G10 for 21 time points; (b)Time series Gene Expression Dataset for 21 by 5 time points
A. Granger Causality test
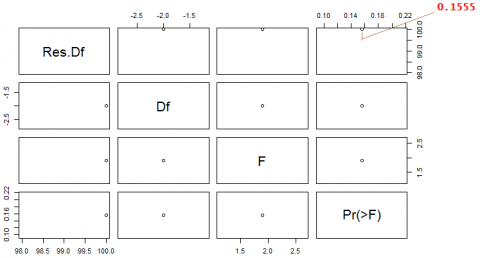
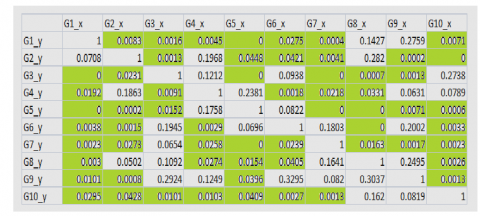
The given dataset is having multiple time series for same set of genes and their expression values are varying with time. To understand the relationship between them, the Granger causality test conducted first with two genes G1 and G2 shown in Figure 10a and then the same test applied to all genes from G1 to G10 shown in Figure 10c.This test help us to know the relationship between one gene with other genes. If the G1 (t) Granger cause G2 (t) then the past value of Gene1 will be helpful to predict the future values of Gene2. The result of Granger causality test shown in Figure 10c, the values highlighted with green color are significant values. The significant value, alpha should be less than 0.05 and all these highlighted green values are significant values. The G1_y Granger causes G2_x as the value is significant level value. And also from Figure 10a where we tested Granger causality between only two genes G1 and G2 that also proved G1 Granger causes G2, as the value obtained is 0.4025 which is less than the significant level value, alpha 0.05. And if we see both the Figure 10b and Figure 10cG2 is not Granger cause G1 as value is greater than significant level in Figure 10b the Granger causality test between G2 and G1 giving 0.1555 and in Figure 10c it is 0.0708.
In the figure the significant level value is 0.425, we accept the null hypothesis as p value is 0.4025>>0.05, this is not significant.
(a)
(b)
(c)
Figure 10. (a) Granger causality test between G1 and G2; (b) Granger causality test between G2 and G1; (c) Granger causality from G1 to G10 genes
The Granger causality test applied on all genes shown in Figure 10c. In Figure 9c, we accept the null hypothesis if p value is 0.1555>0.05, this is not significant.
B. Analyzing the causal relationships using Clustering
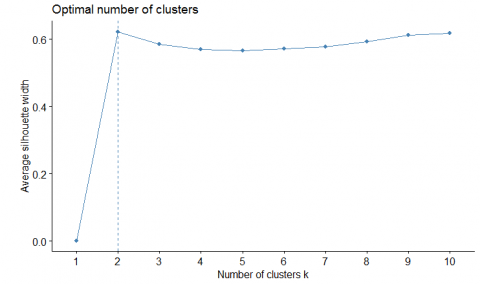
The dataset is also analyzed using k-means clustering. The silhouette method is used to get the best number of clusters for the dataset is obtained as 2 are shown in Figure 11a.
To analyze the causal relationship between the pair of genes the correlation matrix is obtained using k-means clustering is shown in Figure 11b.
In Figure 11b some entries are blank they indicate non-existence of causal relationships of the gene with other genes. The negative values indicate the genes are influenced by other genes and positive values indicate the gene is having correlation with other genes, for example in row Gene G1 we say Gene G1 is more influenced by G5.
(a)
(b)
Figure 11. (a) Optimal value of k, number of clusters; (b) Correlation matrix to estimate the relationship among the genes
The different models for Gene Regulatory inference are used in the paper by analyzing the values associated with each gene with varying time. The Granger causality test is useful in prediction of future values of a gene from the past values of another gene. It also helps us to predict which genes are not affected in future by past values, and through this test the causal relationship between every gene with other genes in the time varying gene expression dataset. The clustering technique groups the genes into different cluster having close correlation with other genes. The insilico network of size 10 for the GRN inference is considered, in future the GRN inference will be performed on large size network structure of complex genetic disease.
[1] Barman, S., Kwon, Y.K. (2018). A Boolean network inference from time-series gene expression data using a genetic algorithm. Bioinformatics, 34(17): i927-i933. https://doi.org/10.1093/bioinformatics/bty584
[2] Barman, S., Kwon, Y.K. (2017). A novel mutual information-based Boolean network inference method from time-series gene expression data. PloS One, 12(2): e0171097. https://doi.org/10.1371/journal.pone.0171097
[3] Yang, Y., Lichtenwalter, R.N., Chawla, N.V. (2015). Evaluating link prediction methods. Knowledge and Information Systems, 45(3): 751-782. https://doi.org/10.1007/s10115-014-0789-0
[4] Ahmed, S.S., Roy, S., Kalita, J. (2018). Assessing the effectiveness of causality inference methods for gene regulatory networks. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 17(1): 56-70. https://doi.org/10.1109/TCBB.2018.2853728
[5] Peignier, S., Schmitt, P., Calevro, F. (2021). Data-driven gene regulatory networks inference based on classification algorithms. International Journal on Artificial Intelligence Tools, 30(4): 2150022. https://doi.org/10.1142/S0218213021500226
[6] Mao, L., Resat, H. (2004). Probabilistic representation of gene regulatory networks. Bioinformatics, 20(14): 2258-2269. https://doi.org/10.1093/bioinformatics/bth236
[7] Zhao, H., Duan, Z.H. (2019). Cancer genetic network inference using gaussian graphical models. Bioinformatics and Biology Insights, 13. https://doi.org/10.1177/1177932219839402
[8] Wang, R.S. (2013). Ordinary differential equation (ODE), model. Encyclopedia of Systems Biology. In: Dubitzky W., Wolkenhauer O., Cho KH., Yokota H. (eds) Encyclopedia of Systems Biology. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-9863-7_381
[9] Sakamoto, E., Iba, H. (2001). Inferring a system of differential equations for a gene regulatory network by using genetic programming. In Proceedings of the 2001 Congress on Evolutionary Computation (IEEE Cat. No. 01TH8546), 1: 720-726. https://doi.org/10.1109/CEC.2001.934462
[10] Kordmahalleh, M.M., Sefidmazgi, M.G., Harrison, S.H., Homaifar, A. (2017). Identifying time-delayed gene regulatory networks via an evolvable hierarchical recurrent neural network. BioData Mining, 10(1): 1-25. https://doi.org/10.1186/s13040-017-0146-4
[11] Meyer-Baese, A., Schmid, V.J. (2014). Pattern Recognition and Signal Analysis in Medical Imaging. Elsevier. https://doi.org/10.1016/C2012-0-00347-X
[12] Ghadle, S., Tripathi, R., Kumar, S., Munde, V. (2021). Study on analysis of gene expression dataset and identification of differentially expressed genes. In Intelligent Computing and Networking, 146: 253-259. https://doi.org/10.1007/978-981-15-7421-4_23