Theoretical Investigation of Structural and Optoelectronic Properties of Ternary Acetylides A2MC2 (A = Li, Na, K) and (M = Te, Pb, Pt)

Abdelhamid Badaoui* | Maamar Belhadji | Houari Ameur | Noureddine Kaid
© 2020 IIETA. This article is published by IIETA and is licensed under the CC BY 4.0 license (http://creativecommons.org/licenses/by/4.0/).
OPEN ACCESS
In this work, the structural and optoelectronic parameters of the ternary bialkali acetylides A2MC2 (A = Li, Na, K) and (M = Te, Pd, Pt) with trigonal structures are investigated. The study is conducted by using the first-principles pseudo-potential plane-wave technique, which is based on the Density Functional Theory (DFT) and integrated in the CASTEP software. A special attention is paid to the Tellurium based compounds, since they are less studied. A satisfactory agreement is reported between our results of the structural parameters and those of the literature. Predictions are performed to determine the values of the band gaps and optical parameters.
theoretical study, density functional theory, optoelectronic properties, material engineering, material characterization, ternary acetylides
The ternary bialkali acetylides A2MC2 are very interesting materials. During few decades, many research works have been conducted on these materials in the field of inorganic chemistry [1-5]. In general, these materials may be synthetized by employing solid-state reaction of parent A2C2 with considered metal M at high temperature [6-8]. Many compounds were synthetized involving Rb, K, Na, and Cs alkali metals and Pt and Pd transition metals [1, 2, 4, 7, 9-11]. Terdik et al. [12] predicted that the photo-emissive properties of Cs2Te may be enhanced by adding an acetylenic unit to obtain the Cs2TeC2 compound. Therefore, related materials like the Li2TeC2 may have the same photo-emissive characteristics. Recently, tellurium based ternary acetylides Li2TeC2 (Na2TeC2) were synthetized for the first time by Németh et al. [13]. They obtained these compounds after synthetizing Li2C2 (Na2C2) by reacting the elemental Li (Na) with acetylene gas. New materials with distinct structures and different properties have been updated by Głodek et al. [14], Chan et al. [15], Svahn et al. [16], Marmol et al. [17], and Sanchez-de-Diego et al. [18], using multiple diagnostic techniques. On the other hand, there are many fields of use of these structures in several areas, as shown by Lankelma et al. [19], Tian et al. [20], Kuroda et al. [21], Hau et al. [22], and Zhou et al. [23].
The aim of this work is to inspect the structural and optoelectronic characteristics of the A2MC2 (A = Li, Na, K) and (M = Te, Pb, Pt) family. The study is based on the density functional theory. The CASTEP software is used as a tool to perform the calculations.
The first-principles pseudo-potential plane-wave technique, which is based on the Density Functional Theory (DFT) and integrated in the CASTEP software was used in our calculations [24].
The generalized gradient approximation of Perdew et al. [25] was utilized to ensure the exchange-correlation functional. The Monkhorst-Pack technique [26] was adopted to integrate the special points sampling over the Brillouin zone. Meshes with 8×8×8 k-point were considered. 360 eV was the energy selected value of cut-off. The minimization approach of Broyden-Fletcher-Golfarb-Shanno (BFGS) was utilized to determine the structural parameters [27].
4.1 Structural characteristics
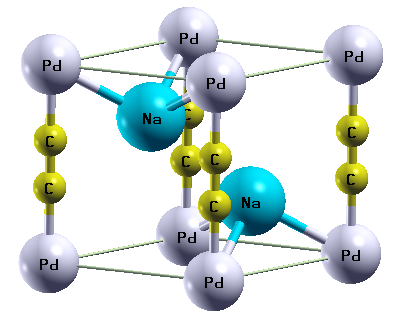
The ternary acetylides compounds A2MC2 are known to crystallize in a trigonal structure with space group (P3m1, 164) (Figure 1). These compounds are characterized by atomic fractional coordinates (zA, zC) and lattice parameters (a, c).
Figure 1. Trigonal structure of ternary acetylides compounds A2MC2
Table 1. (a and c), B, and Eg for the A2MC2 compounds
|
|
Lattice constant ɑ (Å) |
Lattice constant c (Å) |
Bulk Modulus B (GPɑ) |
Energy Gap Eg (eV) |
||||
|
|
This work |
Exp. |
This work |
Exp. |
This work |
Exp. |
This work |
Exp. |
|
Li2TeC2 |
4.25 |
- |
6.00 |
- |
45.12 |
- |
1.448 |
- |
|
Li2PdC2 |
3.85 |
- |
5.30 |
- |
11.28 |
- |
0.243 |
- |
|
Li2PtC2 |
3.98 |
- |
5.26 |
- |
44.68 |
- |
0.094 |
- |
|
Na2TeC2 |
4.79 |
- |
6.16 |
- |
9.69 |
- |
1.737 |
- |
|
Na2PdC2 |
4.55 |
4.4641 |
5.32 |
5.2661 |
26.89 |
- |
0.997 |
- |
|
Na2PtC2 |
4.64 |
4.5031 |
5.27 |
5.2051 |
27.14 |
- |
0.507 |
- |
|
K2TeC2 |
5.19 |
- |
6.11 |
- |
26.85 |
- |
2.244 |
- |
|
K2PdC2 |
5.13 |
5.1052 |
5.32 |
5.2822 |
23.12 |
- |
1.591 |
- |
|
K2PtC2 |
5.18 |
5.1232 |
5.28 |
5.2182 |
65.81 |
- |
1.129 |
- |
Note: (1): Ref. [6]; (2): Ref. [8].
Table 1 summarises the values of the parameters a and c. It reports a comparison between our findings and the available experimental data. As clearly remarked, both results are in good agreement. In the knowledge of authors, no experimental results for the bulk modulus or its pressure derivative are available in the literature.
4.2 Electronic characteristics
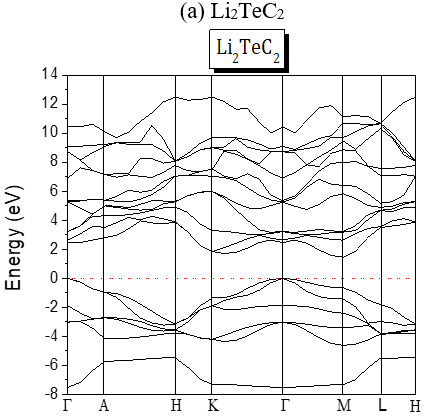
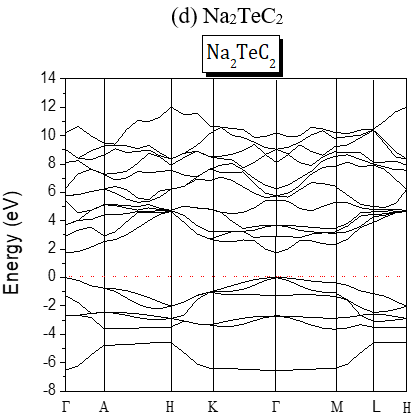
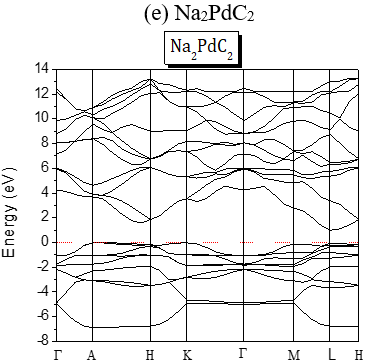
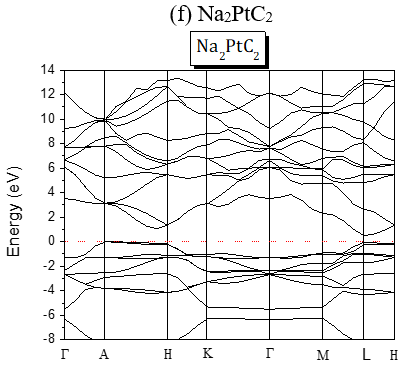
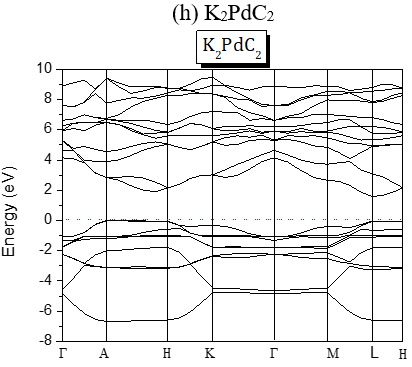
The structures of the energy band of the investigated compounds are shown in Figure 2. Our results are predictions, since no experimental details are found in the literature for the band gap of these compounds. It is seen from Figure 2 that for Te-based compounds, Na2TeC2 and K2TeC2 present G-G direct gaps of 1.737 and 2.244 eV respectively while Li2TeC2 is G-M indirect gap of 1.448 eV.
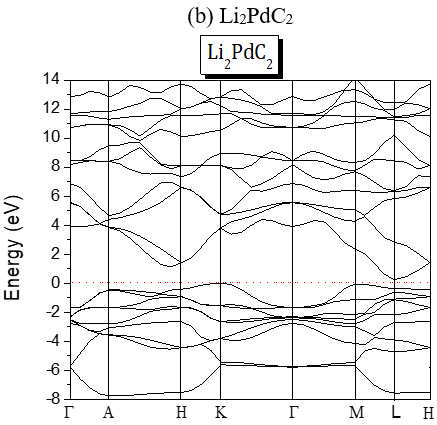
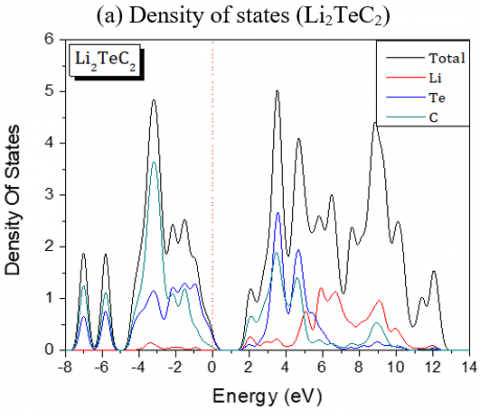
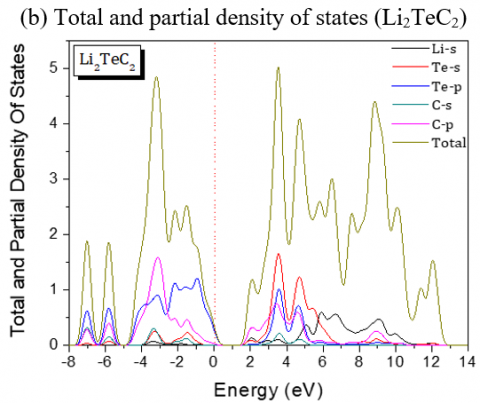
The band gaps are all A-L indirect except for Li2PdC2 which is K-L one. The partial and global density of states of Li2TeC2 and Li2PdC2 are shown in Figure 3, which reveals the existence of upper valence bands consisting of Te-p and C-p orbitals located before the Fermi level for Li2TeC2, while for Li2PdC2 the orbitals are Pd-p and C-p. The same behavior is seen for the others but the upper valence bands consist of Pd-d or Pt-d and C-p orbitals. For the latters, the strong hybridization between Pd-d or Pt-d and C-p states in the upper valence band shows a covalent bonding character.
Figure 2. Band Structures of the investigated A2MC2 compounds
Figure 3. Partial and global density of states of Li2TeC2 and Li2PdC2 compounds
4.3 Optical properties
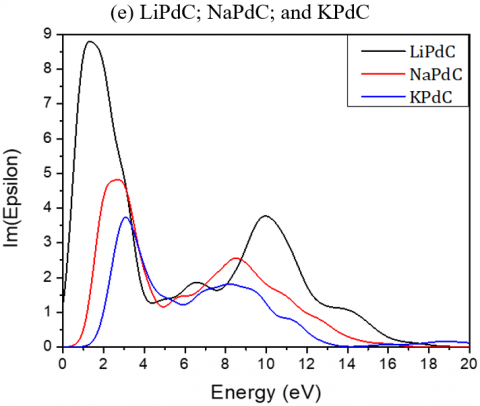
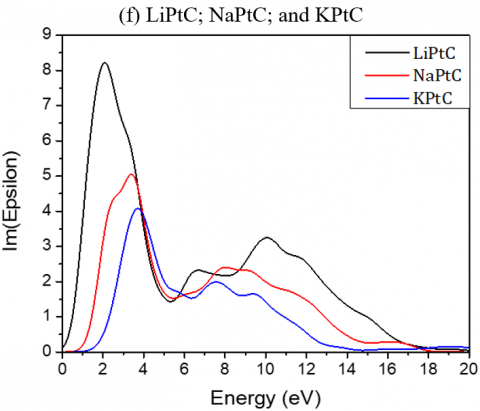
The real (dispersive) and imaginary (absorptive) parts of the dielectric functions for the investigated compounds as function of the photon energy are shown in Figure 4.
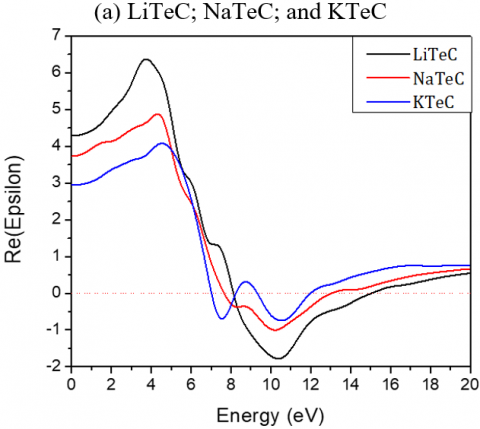
The static dielectric constants ε (0) for Li2TeC2, Na2TeC2 and K2TeC2 are respectively 4.28, 3.73, and 2.94 and reach the peaks at about 3.73, 4.32 and 4.5 eV. For Li2PdC2, Na2PdC2 and K2PdC2, the ε (0) values are 16.62, 5.19, and 3.42 respectively.
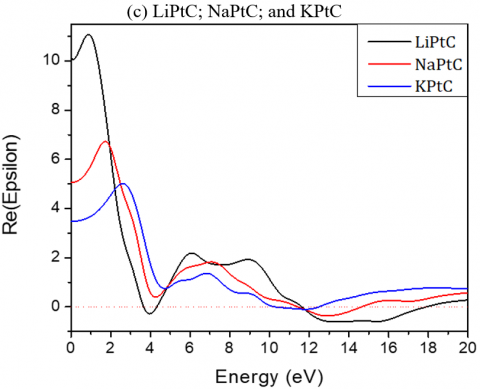
While for Li2PtC2, Na2PtC2 and K2PtC2, the static dielectric constants ε (0) are respectively 10.17, 5.07, and 3.48 eV and reach their maxima at 0.88, 1.72 and 2.61 eV.
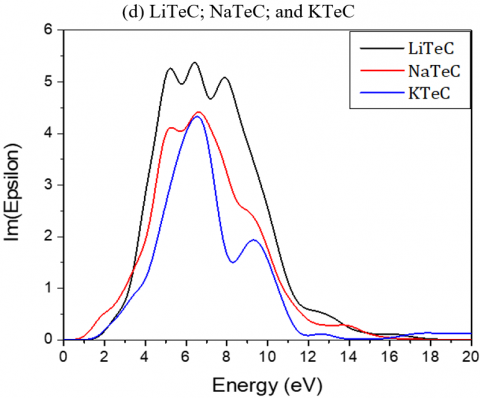
The absorptive part is mainly characterized by the broad peak. The transitions from the valence to the conduction bands are given by the peaks of the dielectric functions.
The maximum mounts of the peak for Li2TeC2, Na2TeC2 and K2TeC2 are respectively at 6.43, 6.62, and 6.54 eV.
For Li2PdC2, Na2PdC2 and K2PdC2, the peaks are at 1.32, 2.70 and 3.10 eV respectively, while for Li2PtC2, Na2PtC2 and K2PtC2, they are respectively at 2.08, 5.07, and 3.40 eV.
Figure 4. Imaginary and real parts of the dielectric function of the investigated A2MC2 compounds
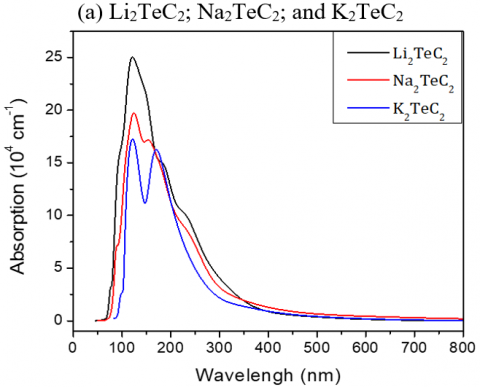
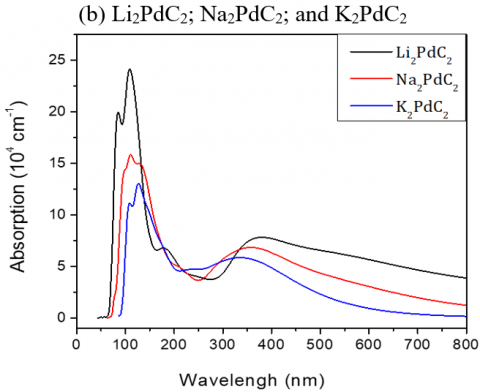
Figure 5 illustrates the absorption spectra of the studied materials, which are relatively similar and present significant absorption bands between 60 and 150 nm, but for Te-based compounds the absorption band is shifted up to 250nm. When plotting the absorption spectra of the studied compounds versus energy, we noticed that optical absorption of Li-based materials starts respectively from very low E.
Note that:
(i) a maximum absorption corresponds to a maximum conduction and to a minimum dispersion,
(ii) a minimum value of the real part of the dielectric function.
Figure 5. Absorption spectra of the investigated A2MC2 compounds
The opto-electronic and structural, characteristics of Te, Pd and Pt based alkali acetylides namely A2MC2 (A = Li, Na, K) and (M = Te, Pd, Pt).
The investigation has been carried out by utilizing the pseudo-potential plane-wave technique that is based on the theory of the density functional density. The generalized gradient approximation has been also adopted. The structural parameters were in satisfactory agreement with the available experimental findings. The energy gaps and optical parameters are predicted. The Te-based compounds present higher gap values. The investigated materials also show band absorption situated in the ultra-violet region.
[1] Ruschewitz, U. (2006). Ternary alkali metal transition metal acetylides. Zeitschrift für Anorganische und Allgemeine Chemie, 632(5): 705-719. https://doi.org/10.1002/zaac.200600017
[2] Buschbeck, R., Low, P.J., Lang, H. (2011). Homoleptic transition metal acetylides. Coordination Chemistry Reviews, 255(1-2): 241-272. https://doi.org/10.1016/j.ccr.2010.07.004
[3] Ruschewitz, U. (2003). Binary and ternary carbides of alkali and alkaline-earth metals. Coordination Chemistry Reviews, 244(1-2): 115-136. https://doi.org/10.1016/S0010-8545(03)00102-4
[4] Brandsma, L. (2003). Best Synthetic Methods: Acetylenes, Allenes And Cumulenes. Elsevier.
[5] Stang, P.J., Diederich, F. (2008). Modern Acetylene Chemistry. John Wiley & Sons.
[6] Hemmersbach, S., Zibrowius, B., Ruschewitz, U. (1999). Na2C2 und K2C2: Synthese, kristaliistruktur und spektroskopishe eigenschaften. Zeitschrift für Anorganische und Allgemeine Chemie, 625(9): 1440-1446. https://doi.org/10.1002/(SICI)1521-3749(199909)625:9%3C1440::AID-ZAAC1440%3E3.0.CO;2-R
[7] Ruschewitz, U., Müller, P., Kockelmann, W. (2001). Zur kristallstruktur von Rb2C2 und Cs2C2. Zeitschrift für Anorganische und Allgemeine Chemie, 627(3): 513-522. https://doi.org/10.1002/1521-3749(200103)627:3%3C513::AID-ZAAC513%3E3.0.CO;2-I
[8] Moissan,H. (1898). Sur les conditions de formation des carbures alcalins, des carbures alicalno-terreux et du carbure de magnesium. Acad. Sci, 126: 302-308.
[9] Kockelmann, W., Ruschewitz, U. (1999). Novel ternary alkali metal silver acetylides MIAgC2 (Ml = Li, Na, K, Rb, Cs). Angewandte Chemie International Edition, 38(23): 3492-3495. https://doi.org/10.1002/(SICI)1521-3773(19991203)38:23%3C3492::AID-ANIE3492%3E3.0.CO;2-E
[10] Billetter, H., Wallraff, T., Schwarz, U., Smith, R.I., Ruschewitz, U. (2010). Ternary transition metal acetylides Al2M0C2 (Al = K, Rb; M0 = Pd, Pt): Neutron diffraction studies and electronic properties. Zeitschrift für Anorganische und Allgemeine Chemie, 636(9-10): 1834-1838. https://doi.org/10.1002/zaac.201000108
[11] Hamberger, M., Liebig, S., Friedrich, U., Korber, N., Ruschewitz, U. (2012). Evidence of solubility of the acetylide lon C22-: Synteses and crystal structures of K2C2.2NH3, Rb2C2.2NH3, and Cs2C2.7NH3. Angewandte Chemie International Edition, 51(52): 13006-13010. https://doi.org/10.1002/anie.201206349
[12] Terdik, J.Z., Németh, K., Harkay, K.C., Terry Jr, J.H., Spentzouris, L., Velázquez, D., Rosenberg, R., Srajer, G. (2012). Anomalous work function anisotropy in ternary acetylides. Physical Review B, 86(3): 035142. https://doi.org/10.1002/anie.201206349
[13] Németh, K., Unni, A.K., Kalnmals, C., Segre, C.U., Kaduk, J., Bloom, I.D., Maroni, V.A. (2015). The synthesis of ternary acetylides with tellurium: Li2TeC2 and Na2TeC2. RSC Advances, 5(69): 55986-55993. https://doi.org/10.1039/C5RA08983B
[14] Głodek, M., Pawlędzio, S., Makal, A., Plażuk, D. (2019). The impact of crystal packing and aurophilic interactions on the luminescence properties in polymorphs and solvate of aroylacetylide-gold (I) complexes. Chemistry-A European Journal, 25(57): 13131-13145. https://doi.org/10.1002/chem.201901101
[15] Chan, K.T., Tong, G.S.M., To, W.P., Yang, C., Du, L., Phillips, D.L., Che, C.M. (2017). The interplay between fluorescence and phosphorescence with luminescent gold (I) and gold (III) complexes bearing heterocyclicarylacetylide ligands. Chemical Science, 8: 2352-2364. https://doi.org/10.1039/C6SC03775E
[16] Svahn, N., Moro, A.J., Roma-Rodrigues, C., Puttreddy, R., Rissanen, K., Baptista, P.V., Fernandes, A.R., Lima, J.C., Rodríguez, L. (2018). The important role of the nuclearity, rigidity, and solubility of phosphane ligands in the biological activity of gold(I) complexes. Chemistry-A European Journal, 24(55): 14654-14667. https://doi.org/10.1002/chem.201802547
[17] Marmol, I., Virumbrales-Munoz, M., Quero, J., Sanchez-de-Diego, C., Fernandez, L., Ochoa, I., Cerrada, E., Yoldi, M.J.R. (2017). Alkynyl gold (I) complex triggers necroptosis via ROS generation in colorectal carcinoma cells. Journal of Inorganic Biochemistry, 176: 123-133. https://doi.org/10.1016/j.jinorgbio.2017.08.020
[18] Sanchez-de-Diego, C., Marmol, I., Perez, R., Gascon, S., Rodriguez-Yoldi, M.J., Cerrada, E. (2017). The anticancer effect related to disturbances in redox balance on Caco-2 cells caused by an alkynyl gold(I) complex. Journal of Inorganic Biochemistry, 166: 108-121. https://doi.org/10.1016/j.jinorgbio.2016.11.009
[19] Lankelma, M., Vreeken, V., Siegler, M.A., van der Vlugt, J.I. (2019). Dinuclear Gold complexes supported by wide bite angle diphosphines for preorganization-induced selective dual-gold catalysis. Inorganics, 7(3): 28. https://doi.org/10.3390/inorganics7030028
[20] Tian, Z., Yang, X., Liu, B., Zhao, J., Zhong, D., Wu, Y., Zhou, G., Wong, W.Y. (2018). Novel AuI polyynes and their high optical power limiting performances both in solution and in prototype devices. Journal of Materials Chemistry C, 6(22): 6023-6032. https://doi.org/10.1039/C8TC01539B
[21] Kuroda, Y., Nakamura, S., Srinivas, K., Sathyanarayana, A., Prabusankar, G., Hisano, K., Tsutsumi, O. (2019). Thermochemically stable liquid-crystalline gold (I) complexes showing enhanced room temperature phosphorescence. Crystals, 9(5): 227. https://doi.org/10.3390/cryst9050227
[22] Hau, F.K.W., Cheung, K.L., Zhu, N., Yam, V.W.W. (2019). Calixarene-based alkynylbridged gold (I)isocyanide and phosphine complexes as building motifs for the construction of chemosensors and supramolecular architectures. Organic Chemistry Frontiers, 6(8): 1205-1213. https://doi.org/10.1039/C9QO00258H
[23] Zhou, Y.P., Wei, Z.W., Lin, Z.J., Ling, H.T., Guo, Z., Zhang, M., Lam, C.K., Ye, B.H., Chao, H.Y. (2017). Diverse binding of important anions in 1-D tricopper anion coordination polymer (ACP) architectures. CrystEngComm, 19(17): 2349-2358. https://doi.org/10.1039/C7CE00087A
[24] Segall, M., Lindan, P.J., Probert, M.A., Pickard, C.J., Hasnip, P.J., Clark, S., Payne, M. (2002). First-principes simulation: Ideas, illustrations and the CASTEP code. Journal of Physics: Condensed Matter, 14(11): 2717.
[25] Perdew, J.P., Burke, K., Ernzerhof, M. (1996). Generalized gradient approximation made simple. Physical Review Letters, 77: 3865. https://doi.org/10.1103/PhysRevLett.77.3865
[26] Monkhorst, H.J., Pack, J.D. (1976). Special points for brillouin-zone integrations. Physical Review B, 13: 5188. https://doi.org/10.1103/PhysRevB.13.5188
[27] Fischer, T.H., Almlof, J. (1992). General methodes for geometry and wave function optimization. The Journal of Physical Chemistry, 96(24): 9768-9774. https://doi.org/10.1021/j100203a036