A.I.A. Alzahrani | S.U. Hassan | M.H. Nazir | A.Zafar | S.Z.J. Zaidi*
© 2022 IIETA. This article is published by IIETA and is licensed under the CC BY 4.0 license (http://creativecommons.org/licenses/by/4.0/).
OPEN ACCESS
Water oxidation could be helpful to have a generation of useful oxidant known as hydrogen peroxide (H2O2) via electrochemical route. A defined structured of molybdenum oxide based electrode was synthesised by a thermal compression treatment over PV-15 composite coating. The coating was then employed for electrochemical water splitting at constant potential electrolysis while the quantities of peroxide of hydrogen produced was measured in an advanced type of cell assembly. The overall findings showed that the confined values of applied potential for H2O2 production are anodic constant potentials of +0.3 V, +0.4 V and +0.5 V vs. Hg/HgO and synthesised peroxide of hydrogen up to 13 ppm in an advanced type of cell assembly.
PV-15, hydrogen peroxide, Molybdenum oxide based electrodes, water splitting
Hydrogen peroxide seems to be an active compound which converted to oxygen, free ionic species and water. It has been greatly employed for many commercial applications such as the production of compounds of industrial needs, bleaching of pulp, treatment of incineration and municipality waste and degradation of harmful organic species. One of the most valuable usage of the hydrogen peroxide is the reagent for the traditional Fenton's reduction based process for the detoxification of organics via a free radical and stable compounds from water treatment slurry. The production of Fenton's based method is depicted by the reaction between peroxide of hydrogen and Fe (II) salts, which produced the hydroxyl based free species over a oxidative potential of $2.33$ $\mathrm{V}$ vs. NHE [1], which can then be employed to oxidized municipality based organic water from waste incineration sources.
Carbon foam is utilized as a cathode for the electrosynthesis of hydrogen peroxide in a divided electrochemical assembly [2]. This electrochemical route provides a good support over the current commercial ways of synthesis by anthraquinone conversion, like its suitable cost and the tendency to develop the specie for degradation over the site of production [3].
Most of the organic molecules were degraded by peroxide of hydrogen by the reduction of dissolved air has been reported as a prospective method via Fenton oxidation [4]. One of the other manageable method for the electrochemical synthesis of peroxide of hydrogen is by 2 electron water oxidation as reflected by equation 1.
$2 \mathrm{H}_2 \mathrm{O} \rightarrow \mathrm{H}_2 \mathrm{O}_2+2 \mathrm{H}^{+}+2 \mathrm{e}^{-} \quad E^0=1.76 \mathrm{~V} v s . \mathrm{NHE}$ (1)
This electrochemical reaction sacrifice with the secondary 4 electrons oxidative oxygen evolution reaction as depicted in equation (2) and the generation of the hydroxyl based ●OH specie which transforms single charge transfer electronic oxidation reaction as shown in equation (3). Most of the reports in updated literature reflects over the oxidative oxygen evolution reaction and the ●OH specie formation with minor focus on the precedence of H2O2 electro-generation [5].
$2 \mathrm{H}_2 \mathrm{O} \rightarrow \mathrm{O}_2+4 \mathrm{H}^{+}+4 \mathrm{e}^{-} \quad E^{\mathrm{O}}=1.23 \mathrm{~V} v s . \mathrm{NHE}$ (2)
$\mathrm{H}_2 \mathrm{O} \rightarrow \mathrm{O}_2+\mathrm{OH}^{\cdot}+4 \mathrm{e}^{-} \quad E^{\mathrm{O}}=2.38 \mathrm{~V} v s . \mathrm{NHE}$ (3)
Recent reports have also demonstrated that dual-electron water splitting to peroxide of hydrogen might be feasible if a coating with reduced over-potential for water to H2O2 can be useful to advance the electro-generation of peroxide of hydrogen during the oxide evolution reaction (OER) [6]. Some oxides of metal have been employed to promote dual-electron water splitting oxidative reaction, which comprise of oxides of Manganese [7], oxides of Titanium [8] and polymeric electrodes for metal air batteries application[9]. Numerous studies of theoretical nature have also showed that these metallic oxides as being aided, at the theoretical yield of water splitting reaction to peroxide of hydrogen (H2O2) via electrochemical route [10,11].
Here we aim to lodge a novel Mo dioxide based substrate towards water splitting for peroxide generation in an undivided electrochemical assembly.
The materials employed are: graphite plates PV-15 from SGL group, Carbon black from Cabot, Reagent grade molybdenum (IV) oxide 99% ,Nitric acid and Nafion® based resin binder solution from Sigma Aldrich, and alcohol, Potassium sulphate, acetone from Fischer scientific. All the materials were employed as obtained.
2.1 Synthesis of the Anodic coating
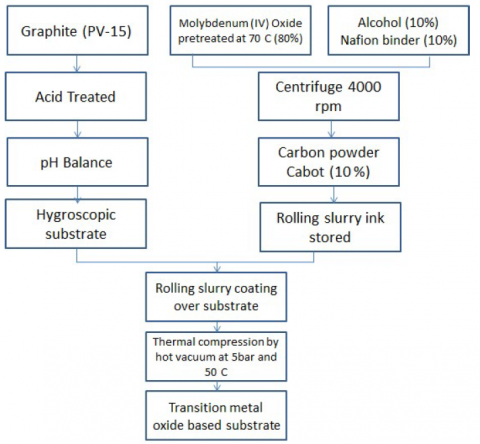
Graphite plates PV-15 was treated with 70% nitric acid at 90 ᴼC for 30 minutes and allowed to rest in deionized water overnight . Slurry of ink was made by mixing molybdenum (IV) oxide 99 % pretreatment at 70 ᴼC with binder and alcohol in 80:10:10. The shear high speed centrifuge was employed at 4000 rpm for 30 minutes. After that, Carbon black with 10% of previous amount was injected in the slurry and centrifuged at 4000 rpm with a diameter of 0.06 cm, 0.10 cm in length wise with PTFE lined, over a magnetic stirrer with cylinder structure. The prepared slurry was coated with brush on both sides of the pretreated PV-15 to form a flat sheet electrode. The electrode was covered with a current collector having stainless steel mesh in a vacuum press to 0.001 cm thickness at 5 bar pressure and 50 ᴼC. The molybdenum (IV) oxide based electrode over PV-15 substrate synthesis method is depicted in Figure 1.
Figure 1. Schematic diagram for the preparation of Molybdenum (IV) oxide coating over PV-15 substrate for the water splitting and subsequent synthesis of hydrogen peroxide
2.2 Electro-synthesis of peroxide
The electro-synthesis studies were conducted by employing an Ivium potentiostat/galvanostat from Alvatech U.K by using its extended Soft Channel 1.
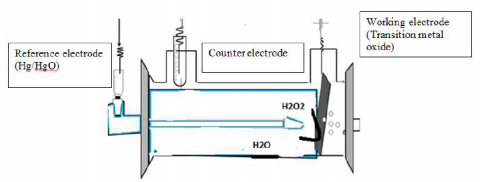
Figure 2. Schematic assembly of advanced type of threeelectrode cell machined with a Molybdenum (IV) oxide coating over PV-15 substrate based working electrode anode, Pt mesh as a counter type electrode with Hg/HgO as reference based electrode connected into a Luggin tube in 150 mL of a 0.5 M K2SO4
The as produced molybdenum (IV) oxide coating was neated with D.I water, purged with nitrogen gas and employed as a working electrode in an advanced type of assembly electrochemical setup as depicted in Figure 2. A 150 cm3 of potassium salt solution with 0.5 mol dm-3 of potassium sulfate was utilized as a background electrolyte for the electrosynthesis of peroxide experiments. A mesh of platinum with (15 mm × 15 mm) was employed as a counter type of electrode while Hg/HgO (0.50 mol dm-3 NaOH) were employed as a reference type of electrode.
2.3 Calculation of Peroxide produced during electrochemical experiments
UV-VIS spectrophotometer was employed to measure the amount of peroxide generated during electrochemical experiments by collecting samples of 0.5 cm3 from exit pathways of cell at notable intervals of experimental time during the electro-synthesis and the concentration was determined by the ammonium metavanadate pathways [10]. The change of phase reaction during the collected specimen colors generated however, the electrolysis was observed with the findings of the conversion of a dull marganda red colour peroxovanadium ionic specie transformed during the chemical change between peroxide present in the sample and metavanadate of ammonium under acidic environment. The absorbance was detected in the UV area of 450 nm radiations.
Under neutral conditions, peroxide was generated by water splitting at the anodic surface as depicted in reaction in equation (1). The electrochemical voltammetry curves represented changes in current densities with reference to the applied constant potential electrolysis. The polarization arc showed in figure 3 determined that after 0.4 V vs. Hg/HgO, the anodic current rises and changes proportionally with the applied constant potential. It is supposedly observed that water splitting to peroxide as shown in reaction 1 carried out at minimum overpotentials and oxidation evolution reaction shown in equation 2 happened primarily after 0.6 V vs. Hg/HgO. As the potential raised towards more positive side oxidative side reaction evolution becomes more obvious.
Figure 3. Voltammetry curve by using molybdenum (IV) oxide based anodic surface in 0.5 M K2SO4 as a background solution potential swept rate = 0.7 mV s-1 with temperature: 298K
For synthesized molybdenum (IV) oxide electrodes under observation, the Faradaic current efficiency (%FCE) can be assumed as the actual electric current density dissipated by the needed electro-synthesis reaction of peroxide production over the total electric current density passed including secondary reactions of oxidative evolutionary reactions [11]. During literature review it was observed that some authors reported peroxide synthesis on the air diffusion electrodes, the determined highest value of 0.003 mol dm–3 at constant potential electrolysis by in the presence of regulated supply of oxygen gas [12]. In similar experiments conducted by researchers peroxide generation was conducted over oxygen diffusion cathodes with the limited provision of oxygen at a current density area of 15 mA cm-2. The peroxide yield was noticed to be increasing trend with a value of 500 mg/L. The employed catalyst was carbon type fine particles over the current metallic support [13]. Evidently, in one of reported studies the electro-synthesis of peroxide was performed in a closed kind of setup which observed more than 700 mg/L of peroxide.
The faradic current efficiency was demonstrated at locative trend of 40% which might be due to evident side reactions in a electrochemical cell assembly [14]. Moreover, in these experiments we showed the synthesis of peroxide without any employment of oxygen or any different gas at molybdenum (IV) oxide based catalyst at a constant potential electrolysis of 0.4 V vs. Hg/HgO.
$\% \mathbf{C E}=\frac{i_{\mathrm{H}_2 \mathrm{O}_2}}{i_{\text {total }}}$ (4)
The tendency anodic constant potential electrolysis on the electro-synthesis of peroxide was measured from 0.2V to 0.7 V vs. Hg/HgO.
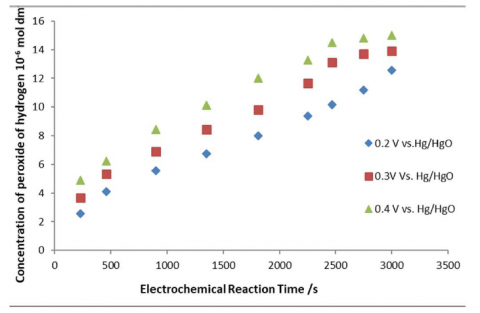
Figure 4. Electro-synthesis of peroxide at 0.2, 0.3 V& 0.4 V vs. Hg/HgO in 0.5 M K2SO4 at molybdenum (IV) oxide based electrode at pH = 7 and Temperature: 298K
Figure 5. Electrolysis at constant applied potential (Chronoamperometry) results for anodic synthesis of peroxide at 0.2,0.3,0.4,0.5 & 0.6 V vs. Hg/HgO in 0.5M K2SO4 at molybdenum (IV) oxide based electrode at pH = 7 and Temperature: 298K
The concentration of peroxide virus electrochemical reaction time was conducted at three different constant applied potentials was showed in Figure 4, while Figures 5 and 6 represented the relevant current densities and %FCE obtained during electrolytic reaction respectively. Peers reported in literature that the peroxide synthesized were dependent and dominated by the side redox reactions which happen to consume more current for a reason to the availability of other competing molecules. The determined sensitivity of more than 50 μA m M−1 cm−2 by provision of a constant potential electrolytic experiments at -0.5 V vs. Ag/AgCl, this actually linked with the observance of other chemical ions which immensely removed out energy which causing current dissipation. The determined peroxide synthesis was only 1.5 μM which reflected the observance of side competing reactions during the reductive reactions at more negative potential electrolytic process [15]. In similar studies, the electro-generation of peroxide was conducted at the expense of energy utilization of 118 kWh kg-1 at an applied current area density of 75 mA cm-2, this phenomenon is more or less a represent the occurrence of predominant side electrochemical changes involved during the electrochemical pathways which nullify over the equilibrium states in the observed reduction reaction transference [16].
Figure 6. Faradic current efficiency obtained for production of hydrogen peroxide at 0.2,0.3 & 0.4 vs. Hg/HgO in 0.5 M K2SO4 at molybdenum (IV) oxide based anode pH = 7 and Temperature 298K.
The studies reflected that peroxide synthesis actually enhance in a trendy way at a constant applied potential of 0.2 V ≤ EA ≤ 0.7 V vs. Hg/HgO. Figure 4 showed that at 0.7 V vs. Hg/HgO, there is a slight increase in the current area density with electro-synthesis reaction time. At potentials greater than 0.6 V vs. Hg/HgO no peroxide was observed and oxidative evolution was evident at the anodic side. Greater EA rooted with the disintegration of peroxide with the presence of secondary oxygen evolution reaction [17].
The overall %FCE is showed in Figure 6 which is very limited for all the electrochemical applied potentials. The rising current efficiency was seen at 0.4 V vs. Hg/HgO but minimize with reaction time for all constant applied potentials electrolysis. However, these observations are corresponded to be novel in lieu of reported experiments as there is no added gas e.g air passage or any kind of specie catalyst were used for peroxide generation at reasonable definitive assembly obtained in electrochemical anodic type of cell.
Here, we have reported that hydrogen peroxide can be electro-synthesized through water splitting using molybdenum (IV) oxide based catalyst in an electrochemical advanced assembly. The peroxide synthesis was observed at 0.3 V vs. Hg/HgO and the quantity of peroxide was increased by rising the potential up to 0.5 V vs. Hg/HgO. Moreover, further rise in constant potential while electro-synthesis has an adverse effect on the electro-generation of peroxide of hydrogen due to the competing side oxidative reactions at increased potentials. This experimental work showed that the electrosynthesis of peroxide can be further enhanced in a more convenient systems with continuous mass transport control. The coating showed in this study represented high stability while performing electrochemical water splitting in an undivided type of cell assembly. These experiments predicted that this method is useful for complex applications of water splitting in the precedence of novel coating surfaces for diversified scientific avenues.
[1] J.M. Noël, A. Latus, C. Lagrost, E. Volanschi, P. Hapiot, J. Am. Chem. Soc., 134 (5) (2012) 2835.
[2] S.Z.J. Zaidi, Y. Luan, C. Harito, L. Utari, B. Yuliarto, F.C. Walsh, Scient. Rep.10 (1) (2020) 1.
[3] S.Z Zaidi, F.C. Walsh, C. Harito, J. Taiwan Inst. Chem E. 104 (2019) 123.
[4] S.Z.J. Zaidi, E. Hurter, F.C. Walsh, C. Ponce de León, Fe (II)-based GDE electrodes for the demineralization of methylene blue dye. Arab.J.Sci.Eng.44 (6) (2019) 5527.
[5] Y. Ando, T. Tanaka, Int. J. Hydrog. Energy. 29 (2004) 1349.
[6] A. Izgorodin, E. Izgorodina, D.R. MacFarlane, Energy Environ. Sci. 5 (2012) 9496.
[7] H. Goto, Y. Hanada, T. Ohno, M. Matsumura, J.Catal. 225 (2004) 223.
[8] K. Fuku, K. Sayama, Chem. Comm. 52 (2016) 5406.
[9] A.F. Alharbi, A.A.M. Abahussain, M.H. Nazir, S.Z.J. Zaidi, Polymers, 14 (2022), 3187.
[10] S. Siahrostami, G.L.Li, V. Viswanathan, J.K. Nørskov, J. Phys. Chem. Lett. 8 (2017) 1157.
[11] R.F.Nogueira, M.C. Oliveira, W.C. Paterlini, Talanta. 66 (2005) 86-91.
[12] R.B. Valim, R.M. Reis, P.S. Castro, A.S. Lima, R.S. R ocha, M. Bertotti, M.R.V. Lanza, Carbon. 61 (2013) 236.
[13] X. Yu, M. Zhou, G. Ren, L. Ma Chem. Eng. J. 263 (2015) 92.
[14] L. Liang, Y. An, M. Zhou, F. Yu, M. Liu, G. Ren, J. Environ. Chem. Eng. 4 (2016) 4400.
[15] M.Y. Hua, H.C. Chen, R.Y. Tsai, C.S. Lai, Talanta, 85 (2011), 631.
[16] J. Moreira, V. Bocalon Lima, L. Athie Goulart, M.R.V. Lanza Appl. Catal. B Environ. 248 (2019) 95.
[17] C. McDonnell-Worth, D.R. MacFarlane, RSC Adv. 4 (2014) 30551.