Amel Bouakkadia* | Youssouf Driouche | Noureddine Kertiou | Djelloul Messadi
© 2020 IIETA. This article is published by IIETA and is licensed under the CC BY 4.0 license (http://creativecommons.org/licenses/by/4.0/).
OPEN ACCESS
Pesticide use in agriculture can cause undesirable effects on humans and the natural environment. Physicochemical properties of pesticides play an important role in determining its distribution and fate in the environment. Chemometric methods can be used to describe how the physicochemical properties vary according to the characteristics of the molecular structure expressed in terms of appropriate molecular descriptors. Quantitative Structure-Property Relationship (QSPR) models can also provide a general overview of the molecular structure that influences these properties. Henry’s law constant (H) is an important property for predicting the solubility and vapor- liquid equilibrium of pesticides. Genetic algorithm/ multi- linear hybrid approach was used to model the log H of 48 pesticides belonging to four chemical classes: ureas, triazines, carbamates and aryloxyalkanoic acids. The 5 explanatory variables model selected is robust and has good fitness and good predictive ability. Two influential points which reinforce the model and an outlier were highlighted. The model can be used to predict the Henry’s law constant of pesticides falling in the applicability domain of our model.
Henry constant, pesticides, molecular descriptors, hybrid QSPR model
Use the quantities increasingly important of phytosanitary products, especially in agriculture, has led many researchers and many managers of the quality of the environment to ask questions about the impact that these products could have on the quality of surface waters [1].
Three variables: aqueous solubility, vapor pressure and Henry's constant, play an essential role in the behavior of pesticides. They can be determinative on how the pesticides will migrate, and therefore fits on the consequences of contamination.
Different models predict these important environmental parameters. Incremental methods are based on structural characteristics such as the type of atom, the type of connection and the local structural environment [2]. The Quantitative Structure-Property Relationship (QSPR) strategies involve physico - chemical properties, such as structural descriptors connectivity indices and descriptors reflecting the electronic structure [3, 4]. Note in addition to the aqueous solubility and Henry's constant, the possibility of using models based on molecular structure and quantum solvation models via the Gibbs solvation ΔGs. The results of these models show substantial differences in the application domains and prediction capabilities [2, 4].
Although QSPR use in predicting Henry’s law constant of pesticides has been rather limited and most of the existing models are derived from very limited data sets [5-7].
According to the literature search, the majority predict the physicochemical properties of a heterogeneous set of pesticides. In our work we studied herbicides that are a homogeneous whole having the same mode of action.
In this study, we applied the methodology QSPR in the hybrid genetic algorithm / multiple linear regression (GA / MLR) approach to predict the Henry constant of 48 pesticides distributed in Table 1 according to their chemical classes: 1-11 (urea), 12-24 (triazines) 25-37 (carbamates), 38-48 (aryloxyalkanoic acids).
The data collected in the literature have been previously separated randomly (SAMPLE command processing software MINITAB data [8]) into a set of calibration for 29 elements for the selection of descriptors by genetic algorithm [9] and the calculation QSPR model , and a validation set of 19 items used for only the external statistical validation.
The goodness of fit and the robustness of the model and its predictive capabilities (internal and external) were examined. Finally, the application domain (AD) was discussed with the Williams diagram (discussed in detail in the researches [10, 11]), which represents the prediction residuals standardized to values leverages (hi).
2.1 MULTIPLE REGRESSION MODEL
Table 1. List of studied compounds: Names, descriptors, observed and prediction values of log H, hi and jackknifed residuals
(•Training o Test)
|
N° |
Composés |
log P |
masse |
ESpm15d |
HATS4v |
R7p+ |
Yobs |
Ypréd |
hi |
eiStd |
|
1 |
Siduron o |
2.86 |
232.33 |
19.106 |
0.108 |
0.011 |
-3.9665 |
-4.38 |
0.084 |
-0.37 |
|
2 |
Chloroxuron • |
3.06 |
290.75 |
19.559 |
0.102 |
0.012 |
-5.7986 |
-4.87 |
0.068 |
0.89 |
|
3 |
Tebuthiuron o |
2.76 |
228.31 |
20.042 |
0.115 |
0.008 |
-6.1636 |
-4.72 |
0.17 |
1.36 |
|
4 |
methabenzthiazuron • |
1.89 |
221.28 |
19.65 |
0.075 |
0.011 |
-7.1958 |
-5.13 |
0.149 |
2.27 |
|
5 |
monolinuron • |
1.81 |
214.65 |
22.128 |
0.274 |
0.028 |
-2.8013 |
-2.47 |
0.373 |
0.57 |
|
6 |
fluometuron • |
2 |
232.21 |
22.479 |
0.109 |
0.011 |
-3.068 |
-4.13 |
0.164 |
-1.2 |
|
7 |
diuron • |
2.15 |
233.1 |
19.658 |
0.129 |
0.02 |
-4.2676 |
-3.6 |
0.059 |
0.63 |
|
8 |
difenoxuron • |
2.29 |
286.33 |
19.547 |
0.085 |
0.006 |
-6.8927 |
-7.27 |
0.095 |
-0.38 |
|
9 |
Linuron o |
2.33 |
249.1 |
22.141 |
0.109 |
0.022 |
-2.9318 |
-1.66 |
0.089 |
1.15 |
|
10 |
methazol • |
2.94 |
261.06 |
22.835 |
0.141 |
0.019 |
-1.5301 |
-1.57 |
0.081 |
-0.04 |
|
11 |
rimsulfuron • |
1 |
431.44 |
25.109 |
0.088 |
0.007 |
-10.6595 |
-9.89 |
0.504 |
1.9 |
|
12 |
Metamitron o |
0.67 |
202.22 |
20.01 |
0.154 |
0.013 |
-6.2269 |
-7.18 |
0.482 |
-1.15 |
|
13 |
Metribuzino |
1.33 |
214.29 |
22.599 |
0.164 |
0.016 |
-6.7375 |
-4.39 |
0.284 |
2.4 |
|
14 |
Prometryn • |
2.37 |
241.35 |
21.989 |
0.055 |
0.015 |
-2.163 |
-2.38 |
0.099 |
0.66 |
|
15 |
atrazine • |
2.06 |
215.69 |
16.975 |
0.097 |
0.019 |
-3.3439 |
-4.59 |
0.161 |
-1.4 |
|
16 |
ametryn • |
1.96 |
227.33 |
21.988 |
0.05 |
0.017 |
-3.1586 |
-3.95 |
0.149 |
0.99 |
|
17 |
Propazine o |
2.47 |
229.71 |
17.058 |
0.099 |
0.02 |
-3.2204 |
-2.26 |
0.149 |
-0.68 |
|
18 |
Simazine o |
1.65 |
201.66 |
16.885 |
0.062 |
0.018 |
-3.4672 |
-4.81 |
0.272 |
-1.36 |
|
19 |
Terbuthylazine o |
2.14 |
229.71 |
22.003 |
0.06 |
0.016 |
-3.2204 |
-2.34 |
0.107 |
0.8 |
|
20 |
prometon • |
2.03 |
225.29 |
16.849 |
0.055 |
0.016 |
-3.3585 |
-5.08 |
0.217 |
-2.15 |
|
21 |
terbutryn • |
2.04 |
241.35 |
19.201 |
0.229 |
0.034 |
-3.0357 |
-2.34 |
0.3 |
1.03 |
|
22 |
hexazinone • |
2.37 |
252.32 |
20.975 |
0.115 |
0.009 |
-7.3809 |
-5.27 |
0.124 |
2.22 |
|
23 |
Dipropetryn o |
2.72 |
255.38 |
22.242 |
0.046 |
0.01 |
-2.91 |
-2.97 |
0.095 |
-0.05 |
|
24 |
desmetryn • |
1.62 |
213.3 |
21.988 |
0.057 |
0.017 |
-3.2814 |
-2.61 |
0.178 |
0.77 |
|
25 |
desmedipham • |
3.02 |
300.31 |
20.184 |
0.064 |
0.009 |
-5.1567 |
-5.04 |
0.09 |
0.11 |
|
26 |
Carbetamide o |
1.32 |
236.27 |
20.394 |
0.098 |
0.01 |
-4.8356 |
-5.94 |
0.153 |
-1.04 |
|
27 |
chlorpropham • |
2.79 |
213.66 |
19.555 |
0.144 |
0.016 |
-2.5391 |
-3.14 |
0.092 |
-0.6 |
|
28 |
Phenmedipham o |
3.14 |
300.31 |
20.173 |
0.076 |
0.008 |
-5.2365 |
-5.21 |
0.085 |
0.02 |
|
29 |
Pebulate o |
2.79 |
203.34 |
23.186 |
0.055 |
0.01 |
0.3159 |
-1.22 |
0.182 |
-1.47 |
|
30 |
prosulfocarb • |
3.83 |
251.39 |
23.235 |
0.064 |
0.007 |
-1.0231 |
-13.28 |
0.186 |
-0.31 |
|
31 |
EPTC o |
2.39 |
189.32 |
23.18 |
0.051 |
0.014 |
0.1931 |
-0.65 |
0.189 |
-0.81 |
|
32 |
cycloate • |
2.7 |
215.35 |
23.193 |
0.106 |
0.011 |
0.0863 |
-2.07 |
0.175 |
-2.49 |
|
33 |
butylate • |
3.2 |
217.37 |
23.181 |
0.07 |
0.015 |
0.4393 |
0.1 |
0.112 |
-0.35 |
|
34 |
vernolate • |
2.86 |
203.34 |
22.113 |
0.217 |
0.027 |
0.3159 |
0.13 |
0.181 |
-0.21 |
|
35 |
thiobencarb • |
3.41 |
257.78 |
23.519 |
0.068 |
0.017 |
-1.4001 |
0.17 |
0.164 |
1.77 |
|
36 |
tri allate • |
3.16 |
304.66 |
23.272 |
0.066 |
0.015 |
-0.2418 |
-1.95 |
0.196 |
-2.04 |
|
37 |
vinclozolin • |
3.4 |
286.11 |
23.486 |
0.152 |
0.016 |
-0.6216 |
-1.82 |
0.168 |
-1.36 |
|
38 |
2,4-D • |
2.37 |
221.04 |
19.556 |
0.168 |
0.022 |
-3.0301 |
-3.02 |
0.081 |
0.01 |
|
39 |
2,4-DB o |
2.8 |
249.09 |
19.456 |
0.145 |
0.022 |
-3.6345 |
-0.69 |
0.084 |
0.85 |
|
40 |
2,4-D-dimethylammonium • |
4.61 |
294.18 |
9.785 |
0.106 |
0 |
-10.8326 |
-10.11 |
0.666 |
3.23 |
|
41 |
2,4,5-T • |
2.89 |
255.48 |
19.737 |
0.181 |
0.022 |
-3.16 |
-2.98 |
0.105 |
0.18 |
|
42 |
Dichlorprop o |
2.91 |
235.07 |
20.063 |
0.206 |
0.026 |
-2.91 |
-1.7 |
0.147 |
1.13 |
|
43 |
triclopyr triethylammonium • |
2.98 |
357.66 |
17.285 |
0.079 |
0.003 |
-9.26 |
-9.54 |
0.196 |
-0.34 |
|
44 |
Fluroxypyr meptyl o |
4.41 |
367.25 |
20.176 |
0.104 |
0.011 |
-4.97 |
-4.23 |
0.482 |
0.89 |
|
45 |
dichlorprop p o |
2.91 |
235.07 |
20.063 |
0.206 |
0.026 |
-2.91 |
-1.7 |
0.147 |
1.13 |
|
46 |
2,4-D-methyl • |
2.4 |
235.07 |
19.798 |
0.144 |
0.031 |
-0.52 |
-0.85 |
0.281 |
-0.46 |
|
47 |
Triclopyr o |
2.65 |
256.47 |
19.971 |
0.096 |
0.014 |
-3.28 |
-4.04 |
0.039 |
-0.67 |
|
48 |
2,4,5-T-trolamine • |
-0.46 |
404.67 |
15.607 |
0.127 |
0.007 |
-16.44 |
-17.34 |
0.589 |
-2.91 |
A multiple regression model between explained variable y and p explicative variables x1, …, xp is written for all i = 1, ..., n:
$\mathbf{y}_{\mathrm{i}}=\beta_{0}+\sum_{\mathrm{j}=1}^{\mathrm{p}} \beta_{\mathrm{j}} \mathbf{x}_{\mathrm{i}}+\varepsilon_{\mathrm{i}}$ (1)
where, yi, xi1, xi2, ..., xip are given respectively on variables y, x1, ..., xp.
βj coefficients are calculated using the method of ordinary least squares. Random variables εi represent the unobservable error terms of the model. These errors can be estimated by ordinary residuals ei, differences between the observed variables yi and the estimated values ŷi.
2.2 Genetic algorithm
The modeling the genetic process has initiated the development of genetic algorithms, which can be exploited in a variety of optimization problems [12]. In this case, a potential solution is considered as an individual in a population. The value of the cost function associated with a measurement solution "adaptation" of the individual associated with its environment. A genetic algorithm simulates the evolution, over several generations, an initial population whose individuals are poorly adapted using genetic operators of reproduction and mutation. After a number of generations, the population consists of well adapted individuals, i.e. the supposed "good" solutions to the optimization problem.
2.3 Calculation and selection of molecular descriptors
We used molecular modeling software Hyperchem 6.03 [13] to represent the molecules and then, using the semi-empirical AM1 method [14] to obtain the final geometry. All calculations were carried out under the RHF formalism [15] without configuration interaction. Molecular structures were optimized using the Polak - Ribiere algorithm for criterion with a root mean square gradient of 0.001 kcal / mol. And optimized geometries were transferred to the Dragon computer software Version 5.3 [16] to calculate 1201 descriptors belonging to different classes. Using the corresponding options DRAGON software, we first eliminated descriptors constant values (standard deviations less than 0.0001), which provide no information, and then those who are highly correlated (R ≥ 0.95) that convey redundant information. For each pair of correlated descriptors is eliminated automatically the one with the highest cross-correlation with other descriptors.
By operating on the calibration data, the sub- descriptor sets were selected by genetic algorithm in the MOBYDIGS release of Todischini [17] maximizing the prediction coefficient $Q_{LOO}^{2}$.
To avoid models with collinearity problems, and no real predictive power, we applied the rule QUIK [18] based on the multivariate index K [19] correlation, this rule is derived from the assumption that the correlation total in the group consisting of model predictive X plus Y response (Kxy) should always be greater than that measured in only the set of predictors (Kx). The size of the model finally selected is determined by the optimal value of the FIT function Kubinyi [20] for comparing models built on n data with different numbers of variables.
2.4 Development and validation of the model
The multilinear regression analysis was performed with the software MOBY DIGS [17].
Acceptable models are only those with a global correlation of [X + y] block (KXY) greater than the global correlation of the X block (KXX) variable, X being the molecular descriptors and y the response variable. Therefore, when there were models of similar performance, those with higher ∆K (KXY – KXX) were selected and further verified.
In general [21], we reject models that do not satisfy the relation:
$D\left( K \right)={{K}_{xy}}-{{K}_{xx}}>0,05$ (2)
The quality of the model was evaluated by different statistics (R2, R2ajusté, Fischer parameter, F, standard error, s), and the standard deviation calculated for all calibration SDEC [22].
Adjusted R2adj, which is calculated using the following formula:
$R_{a d j}^{2}=1-\left[\left(\frac{n-1}{n-m-1}\right)\left(1-R^{2}\right)\right]$ (3)
n is the number of objects of the set of calibration and m is the number of features of the model. Adjusted R2 is a better measure of the proportion of variance explained in the data.
The F ratio is defined as the ratio of the sum of squares of the model and residual sum of squares, which is a comparison of the variance explained by the model and the residual variance: high values of the ratio F indicate a reliable model.
The cross-validation techniques were used for evaluation of the internal prediction ($Q_{L M O}^{2}$; bootstrap), and robustness ($Q_{L O O}^{2}$; Y-scrambling) [22] of the model.
Cross-validation by leave-one -out [23] is to recalculate the model (n-1) objects, and use the resulting model to predict the value of the dependent variable spread compound. The process is repeated for each of the set of n objects calibration. The sum of squares of prediction errors (designated by the acronym for PRESS "Predictive Residual Sum of Squares") is a measure of the estimated dispersion. It is used to define the prediction coefficient ($Q_{L O O}^{2}$), and the standard deviation of prediction SDEP.
A value > 0.5 is generally regarded as satisfactory, and a value > 0.9 is excellent [10].
In fact, if a high value of Q2 is a necessary condition for a possible high predictive ability of a model, this condition alone is not sufficient. To avoid an overestimation of the predictive ability of the model we also applied the procedure leave- many- out (LMO), excluding 20% of the objects in each stage.
The QSPR, models because (often) their complexity and sophistication of the tools used chemometrics can be a source of accidental correlations. In order to establish that the model is not due to chance, we applied the randomization test of Y (Y- scrambling) [24]. The test consists in generating a vector of the property studied by random permutation of the components of the real vector. We then calculate the vector obtained a QSPR model, as usual. This process is repeated 100 times in this study.
In the bootstrap validation technique simulating new samples of size (n) by random draws with replacement. In this way the calibration assembly, which retains its initial size (n), consists in general of objects repeated, collecting all the evaluation excluded [25, 26] objects. The model is calculated for all of the calibration and predicted responses for all evaluation. All the squares of differences between predicted and actual values of the set of objects are collected in the evaluation PRESS. This construction procedure sets calibration and evaluation is repeated many thousands of times (8000 in this study), the PRESS is added, and a calculated average predictive ability [25].
Application of the model, calculated on the whole calibration of the 19 compounds of the validation set, used to check reliably predictive ability of the model obtained. With R2, and the parameters are then useful SDEPext index (ext) in relation to the objects of the set of external validation or those of the overall evaluation obtained by bootstrapping.
The predictive power of the regression model developed on the selected training set is estimated on the predicted values of prediction set chemicals, by the external Q2 that is defined [27] as:
$Q_{E X T}^{2}=1-\frac{\sum_{i=1}^{n_{e t}}\left(\hat{y}_{i / i}-y_{i}\right)^{2} / n_{E X T}}{\sum_{i=1}^{n_{tr}}\left(y_{i}-\bar{y}_{T r}\right)^{2} / n_{T r}}$ (4)
where, yi and $\hat{y}_{i / i}$ are, respectively, the measured and predicted (over the prediction set) values of the dependent variable, and $\bar{y}_{T r}$ the averaged value of the dependent variable for the training set. ntr and next are the number of training set objects and the number of objects in the external set, respectively.
An additional external validation according to the research [28] is applied solely to the test set. According to the recommended criteria of Tropsha [29], a predictive QSPR model, must attend the following conditions:
$Q_{E x T}^{2}>0.5$ (5)
$R^{2}>0.6$ (6)
$\left(R^{2}-R_{0}^{2}\right) /<0.1$ and $0.85<\mathrm{k}<1.15$ (7)
$\left(R^{2}-R_{0}^{\prime 2}\right) /<0.1$ and $0.85<\mathrm{k}^{\prime}<1.15$ (8)
where,
$R=\frac{\sum\left(y_{i}-\bar{y}\right)\left(\tilde{y}_{i}-\overline{\tilde{y}}\right)}{\sqrt{\sum\left(y_{i}-\bar{y}\right)^{2} \sum\left(\tilde{y}_{i}-\overline{\tilde{y}}\right)^{2}}}$ (9)
$R_{0}^{2}=1-\frac{\sum\left(y_{i}-y_{i}^{r_{0}}\right)^{2}}{\sum\left(y_{i}-\bar{y}\right)^{2}}$ (10)
$R_{0}^{\prime 2}=1-\frac{\sum\left(\tilde{y}_{i}-\tilde{y}_{i}^{r_{0}}\right)^{2}}{\sum\left(\tilde{y}_{i}-\tilde{y}\right)^{2}}$ (11)
and
$\mathrm{k}=\frac{\sum\left(\mathrm{y}_{\mathrm{i}} \tilde{\mathrm{y}}_{\mathrm{i}}\right)}{\sum\left(\tilde{\mathrm{y}}_{\mathrm{i}}\right)^{2}}$ (12)
$\mathrm{k}^{\prime}=\frac{\sum\left(\mathrm{y}_{\mathrm{i}} \tilde{\mathrm{y}}_{\mathrm{i}}\right)}{\sum\left(\mathrm{y}_{\mathrm{i}}\right)^{2}}$ (13)
where, R is the correlation coefficient between the calculated and experimental values in the test set; $R_{0}^{2}$ (calculated versus observed values) and $R_{0}^{'2}$ (observed versus calculated values) are the coefficients of determination; k and k' are slopes of regression lines through the origin of calculated versus observed and observed versus calculated, respectively; $y_{i}^{r_{0}}$ and $\tilde{y}_{i}^{r_{0}}$ are defined as $y_{i}^{r_{0}}=k \tilde{y}$ and $\tilde{y}_{i}^{r_{0}}=k^{\prime} y$, respectively; and the summations are over all samples in the test set.
The reason to use $R_{0}^{2}$ and require k values that are close to 1 is that when actual versus predicted properties are compared, an exact fit is required, not just a correlation.
2.5 Applicability domain
The applicability domain was discussed with Williams diagram representing external studentized residuals (eistd) and hi leverage which are the diagonal elements of the matrix $\left( {\underset{\scriptscriptstyle\thicksim}{H}} \right)$ crossing observed quantities (vectors y) the quantities estimated (vectors).
External studentized residuals are obtained from the relationship:
$e_{i s t d}=e_{i}^{*} \sqrt{\frac{n-p-1}{n-p-\left(e_{i}^{*}\right)^{2}}}$ (14)
where, $e_{i}^{*}$,(internal studentized residue) is the ratio:
$e_{i}^{*}=\frac{e_{i}}{s \sqrt{1-h_{i i}}}$ (15)
s is the standard deviation:
$s=\sqrt{\frac{\sum\left(y_{i}-\hat{y}_{i}\right)^{2}}{n-p}}$ (16)
n is the number of objects of the set of calibration and p is the number of the model parameters (equal to 6 in this work). Eq. (17) defines the leverage of a compound in the original space of the independent variables (xi):
${{h}_{i}}= {{\underset{\scriptscriptstyle\thicksim}{x}}_{i}}{{({{\underset{\scriptscriptstyle\thicksim}{X}}^{T}}\underset{\scriptscriptstyle\thicksim}{X})}^{-1}} \underset{\scriptscriptstyle\thicksim}{x}_{i}^{T}$ (i = 1, 2, ..., n) (17)
where, ${{\underset{\scriptscriptstyle\thicksim}{x}}_{i}}$ is the row vector of the descriptors of component i, and ${{\underset{\scriptscriptstyle\thicksim}{X}}_{i}}$ the matrix of the model derived from the values of the descriptors of all calibration, the exponent T designates the vector ( or matrix ) transposed (e).
The critical value of leverage (h*) is fixed at 3p / n (0.6 in this work). If hi < h*, the probability of agreement between measured and predicted values of component i, is as high as that of the compounds of calibration. Compounds with hi >h* positively influence the model when they belong to the calibration set, but will, in the contrary case, the predicted values doubtful without necessarily be aberrant, the residues can be low.
3.1 Experimental data
The data were collected from literature [29], the Henry’s law constant values (H), mainly estimated as Vapor pressure x Molecular weight / Water solubility, and were compiled in the units of Pa m3/mol. The values presented as the logarithm of H.
The data set was randomly divided into training set of 29 compounds and a test set of 19 compounds.
3.2 Model
The genetic algorithm optimization leads to many models of different sizes. The variation of the function FIT shows that among the descriptors can be connected with log H, a subset of five descriptors will probably best suited for modeling MLR. The five best descriptors are: octanol -water partition (log P), molecular mass (M), the spectral moment of order 15 of the edge adjacency matrix weighted by dipole moment d (ESpm15d), autocorrelation of the topological distance (that is to say the number of links of the shortest path between two atoms ) equal to 4 by the weighted lever atomic van der Waals volume of v (HATS4v), the maximum radius of autocorrelation topological distance 7 weighted by polarizability p ( R7p +). Are found by Todeschini et al. [30] relations possible definition for the calculation of the descriptors.
These five descriptors have some collinearity (Kx = 28.97). However, what is most important is that the difference in the correlation block variables X plus the response Y (kxy) and that of the block X (Kx) is large enough (Δ = kxy - Kx ~ 13) (Table 2).
The model based on these descriptors is for equation:
log H = - 13,2 (±2,292) + 1,644(±0,2109) log P – 0,026 (±0,004) mass + 0,491 (±0,077) ESpm15d –12,094(±5,10) HATS4v +207,77 (±42,39) R7p+(18)
Diagnostic statistics collected in Table 2 allow making comparisons and drawing several conclusions.
The values of R2 and R2adj show the goodness of fit, while the small difference between R2 and Q2LOO information about the robustness of the model is further highly significant (high value of the statistic Fisher F). The close values of SDEC and SDEP mean that the ability of the internal prediction of model is not too dissimilar to his adjustment power.
The small difference between Q2LOO and Q2LMO/20 shows good stability in the internal validation, and validation by bootstrap (Q2boot) confirms at once good internal predictive ability and stability of the model.
External statistical validation (Q2ext; EQMPext) attests to the good predictive ability of the compounds did not participate in the calculation model.
Table 2. Diagnostic statistical model (ntr=29, ntest= 19)
|
Parameters |
Values |
|
R2 |
92,89 |
|
$Q_{L O O}^{2}$ |
87,89 |
|
$Q_{L M O / 20 \%}^{2}$ |
86,74 |
|
$Q_{\text {boot}}^{2}$ |
82,91 |
|
$Q_{\text {ext}}^{2}$ |
90,86 |
|
R2adj |
91,35 |
|
EQMP |
1,304 |
|
EQMC |
0,999 |
|
EQMPext |
1,133 |
|
Kx |
28,97 |
|
Kxy |
41,65 |
|
F |
60,12 |
|
S |
1,122 |
The high absolute t-values shown in Table 3 express that the regression coefficients of the descriptors involved in the MLR model are significantly larger than the standard deviation. The t- probability of a descriptor can describe the statistical significance when combined together within an overall collective QSPR model (i.e. descriptors interactions). Descriptors with t- probability values below 0.05 (95 percent confidence) are usually considered statistically significant in a particular model, which means that their influence on the response variable is not merely by chance [31]. The smaller t- probability suggests the more significant descriptor. The t- probability values of three descriptors are very small, indicating that all of them are highly significant descriptors. Models would not be accepted if they contain descriptors with VIFs above a value of five [32].
Table 3. Characteristics of the selected descriptors in MLR model
|
Descriptor |
Dx |
x |
t- value |
t- probability |
VIF |
|
Constant |
2.292 |
-13.242 |
-5.78 |
0.000 |
|
|
log P |
0.210 |
1.644 |
7.79 |
0.000 |
1.0 |
|
mass |
0.004 |
-0.026 |
-6.19 |
0.000 |
1.5 |
|
ESpm15d |
0.077 |
0.491 |
6.34 |
0.000 |
1.2 |
|
HATS4v |
5.100 |
-12.094 |
-2.37 |
0.026 |
1.8 |
|
R7p+ |
42.39 |
207.77 |
4.90 |
0.000 |
2.6 |
Correlation matrix as shown in Table 4 suggests that these descriptors are weakly correlated with each other. Thus, the model can be regarded as an optimal regression equation.
Table 4. Correlation matrix
|
log cte H |
log P |
mass |
ESpm15d |
HATS4v |
|
|
log P |
0.390 |
|
|
||
|
0.036 |
|
|
|||
|
mass |
-0.712 |
-0.039 |
|
|
|
|
0.000 |
0.839 |
|
|
||
|
ESpm15d |
0.538 |
-0.140 |
-0.203 |
|
|
|
0.003 |
0.468 |
0.292 |
|
|
|
|
HATS4v |
0.158 |
-0.127 |
-0.258 |
0.024 |
|
|
0.412 |
0.511 |
0.177 |
0.903 |
|
|
|
R7p+ |
0.637 |
-0.088 |
-0.556 |
0.308 |
0.630 |
|
0.000 |
0.649 |
0.002 |
0.104 |
0.000 |
The statistical parameters obtained for the test set [33], demonstrate the power of the predictivity of the models, as shown in Table 5.
Table 5. Test set goodness metrics
|
|
Method |
MLR |
|
|
R2 |
92.89% |
|
Training set n= 29 |
Q2 |
87.89% |
|
|
Q2ext |
90.86% |
|
|
RMSE |
1.122 |
|
|
r2 |
65.70% |
|
Validation set n= 19 |
R2CVext |
75.75% |
|
|
(R2-R20)/ R2<0.1 |
-0.517 |
|
|
(R2-R’20)/ R2<0.1 |
-0.493 |
|
0.85 < k< 1.15 |
0.9763 |
|
|
0.85< k’< 1.15 |
0.9284 |
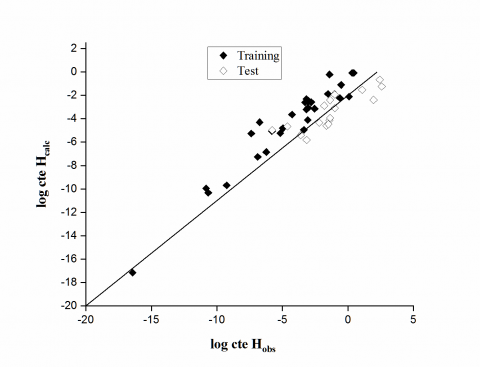
According to the Figure 1 it was clear that the calculated log cte H values were very similar to the experimental values.
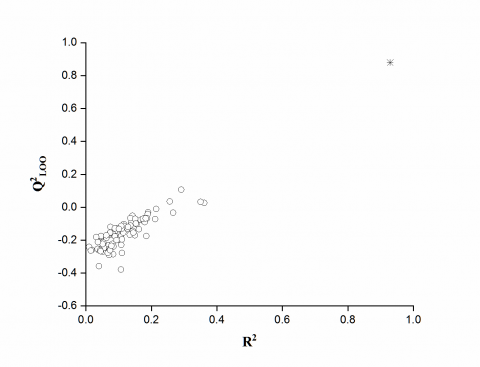
Figure 2 represents the diagram of statistics coefficients Q2LOO and R2 used to compare the results for the randomized models (Circles) in the starting model (star) is the procedure Y - Scrambling. It is clear that the statistics obtained for the modified vectors of the Henry constant (log H) are smaller than those of the actual models; Q2 are lower than 0.10, and most of it even gets Q2 < 0. This ensures that the model established a real basis, and is not due to chance.
Figure 1. Predicted values vs. experimental values for the training, validation sets
Figure 2. Randomization test associated to previous QSPR model. Circles represent the randomly ordered Henry constant, and star corresponds to the real Henry constant
Figure 3. Williams plot of the MLR model for the entire dataset
3.3 Applicability domain
As can be seen from the Williams diagram (Figure 3), 2,4-D- diméthylammonium, has a significant leverage (hi> hi * = 0.62), and it is influential, it reinforces the model.
Additionally, 2,4-D-dimethylammonium having a prediction residual standardized eistd great than 3 standard deviation units is aberrant.
3.4 Interpretation of the model
Henry's constant is a measure of the relative affinity of a compound for the vapor phase and water, and gaseous state being close to the ideal state, H depends primarily on interactions in the aqueous phase.
The values of the coefficients R7p + (207.77), log P (1.644) and ESpm15d (0.491) in equation (18) indicate the order of the positive contribution of these descriptors to the value of H.
The maximum autocorrelation topological distance R, belongs to the class of GETAWAY descriptors [34, 35] R7p+described, to some extent, the size and shape of molecules that with symmetry, play, as we known, a key role in the process of distribution between the two phases. At the same time this descriptor points to the role of dispersion interactions, through the polarizability p.
The partition coefficient n- octanol / water calculated log P (belongs to the class descriptors "Molecular properties") [36], is a mixed solvation descriptor. It reflects both the overall solute interactions with the mass of the surrounding liquid (non- specific effects or macroscopic solvent), and specific binding (usually hydrogen bonds) between the solute and individual solvent molecules (solvent effects specific or microscopic).
The spectral moments of the adjacency matrix of edges (belongs to the class Edegadjency indices) [37, 38] are used to connect the physical (and biological) molecules, directly to their structural components. In addition, by weighting the dipole moment descriptor ESpm15d emphasizes the role of specific interactions in the control of the air / water distribution.
The molecular weight (belongs to the class descriptors constitutional descriptors) is a non-specific structural parameter, easily calculable, which provides information on the size effect of solute molecules. The size of the cavity to be created in the water for receiving the solute molecule, and consequently the energy expended to break the hydrogen bonds necessary for this design, with the increasing size of the molecule. At the same time, this increased size results in increased strength of the forces of interaction and other dispersion [3] capacity. Thus, the overall effect of the mass of the molecules may be minimal or negligible. In our case, the sign of the coefficient of the mass in Eq. (18) indicates that the energy gain due to Coulomb interactions and dispersion outweighs the energy penalty associated with the formation of cavities. Also, with all calibration selected, H varies in opposition to the mass.
HATS4v is another descriptor GETAWAY still emphasizes the role of the size of the molecules, via atomic van der Waals volume, v.
The Henry's Law constants (log H) of 48 pesticides belonging to 4 different chemical classes but having the same mode of action in contrary of other works were randomly separated into two disjoint subsets of elements 29 and 19 respectively. The first was used for the selection, by genetic algorithm, the theoretical molecular descriptors derived from the structure of the molecules (DRAGON software), then the construction of the model, the second set was used for testing.
The multilinear model with five variables presented is robust, with good internal and external predictive capabilities, and a good quality of fit. The 2,4 -D- dimethylammonium, is influential and reinforces the model. Only the 2,4- D- dimethylammonium is aberrant.
With the size, shape and structural component of molecules, the effects of macroscopic and microscopic solvent governing air / water distribution for heterogeneous set of pesticides considered.
The practical utility of the developed QSPR model is to remedy the lack of information on herbicides provided that they belong to the same defined field of application.
[1] Khan, M. (1977). Pesticides in Aquatic Environments. New York: Plenum Press.
[2] Mackay, D., Shiu, W.S., Ma, K.C. (2000). Handbook of Property Estimation Methods for Chemicals: Environmental and Health Sciences. Boethling. R.S., Mackay. D. eds. Lewis, Boca Raton, FL, USA. p. 16.
[3] Dearden, J.C., Schüürmann, G. (2003). Quantitative structure- property relationships for predicting henry's law constant from molecular structure. Environmental Toxicology and Chemistry Journal, 22(8): 1755-1770. https://doi.org/10.1897/01-605
[4] Estrada, E., Delgado, E.J., Alderate, J.B., Jana, G.A. (2004). Quantum- connectivity descriptors in modeling solubility of environmentally important organic compounds. Journal of Computational Chemistry, 25(14): 1787-1796. https://doi.org/10.1002/jcc.20099
[5] English, N.J., Carroll, D.G. (2001). Prediction of Henry’s law constants by a quantitative structure property relationship and neural networks. Journal of Chemical Information and Computer Sciences, 41(5): 1150-1161. https://doi.org/10.1021/ci010361d
[6] Li, A., Doucette, W.J., Andren, A.W. (1994). Estimation of aqueous solubility, octanol/water partition coefficient, and Henry’s law constant for polychlorinated biphenyl’s using UNIFAC. Chemosphere, 29(4): 657-669. https://doi.org/10.1016/0045-6535(94)90037-X
[7] Russell, C.J., Dixon, S.L., Jurs, P.C. (1992). Computer-assisted study of the relationship between molecular structure and Henry’s law constant. Analytical Chemistry, 64(13): 1350-1355. https://doi.org/10.1021/ac00037a009
[8] MINITAB, Release 13.31, Statistical Software, 2000.
[9] Leardi, R., Boggia, R., Terrile, M. (1992). Genetic algorithms as a strategy for feature selection. Journal of Chemometrics, 6(5): 267-281. https://doi.org/10.1002/cem.1180060506
[10] Eriksson, L., Jaworska, J., Worth, A., Cronin, M., Mc Dowell, R.M., Gramatica, P. (2003). Methods for reliability, uncertainty assessment, and applicability evaluations of regression based and classification QSPRs. Environmental Health Perspective Journal, 111(10): 1361-1375. https://doi.org/10.1289/ehp.5758
[11] Tropsha, A., Gramatica, P., Grombar, V.K. (2003). The importance of being Earnest: Validation is the absolute essential for successful application and interpretation of QSPR models. QSAR &Combinatorial Science, 22(1): 69-76. https://doi.org/10.1002/qsar.200390007
[12] Goldberg, D.E. (1989). Genetic Algorithms in Search, Optimization and Machine Learning, Addison Wesley.
[13] HyperchemTM Release 6.03 for Windows, Molecular Modeling System, 2000.
[14] Dewar, M.J.S., Zoebisch, E.G., Ealy, E.F., Stewart, J.J.P. (1985). AMI: A new general purpose quantum mechanical molecular model. Journal of the American Chemical Society, 107(13): 3902-3909. http://doi.org/10.1021/ja00299a024
[15] Levine, I.N. (2000). Quantum Chemistry. 5thed. New Jersey: Prentice Hall.
[16] Todeschini, R., Consonni, V., Dragon, P.M. (2006). Software for the Calculation of Molecular Descriptors. Release 5.3 for windows, Milano.
[17] Todeschini, R., Ballabio, D., Consonni, V., Mauri, A., Pavan, M. (2009). MOBY DIGS software for multilinear regression analysis and variable subset selection by genetic algorithm. Release 1.1 for Windows, Milano.
[18] Todeschini, R., Consonni, V., Maiocchi, A. (1999). The K correlation index: Theory development and its application in chemometrics. Chemometrics and intelligent Laboratory Systems, 46(1): 13-29. https://doi.org/10.1016/S0169-7439(98)00124-5
[19] Todeschini, R. (1997). Data correlation, number of significant principal components and shape of molecules. The K correlation index. Analytica Chimca Acta, 348(1-3): 419-430. https://doi.org/10.1016/S0003-2670(97)00290-0
[20] Kubinyi, H. (1994). Variable selection in QSAR studies: I. An evolutionary algorithm. Quantitative Structure-Activity Relationships Journal, 13(3): 285-294. https://doi.org/10.1002/qsar.19940130306
[21] Todeschini, R., Consonni, V., Mauri, A., Pavan, M. (2004). Detecting “bad” regression models: multicriteria fitness functions in regression analysis. Analytica Chimca Acta, 515(1): 199-208. https://doi.org/10.1016/j.aca.2003.12.010
[22] Draper, N.R., Smith, H. (1998). Applied Regression Analysis. Third Edition. Wiley Series in Probability and Statistics. New York.
[23] Allen, D.M. (1974). The relationship between variable selection and data augmentation and a method for prediction. Technometrics, 16(1): 125-127. https://doi.org/10.2307/1267500
[24] Wold, S., Eriksson, L., Clementi, S. (1995). Statistical validation of QSAR results. In: Van de Waterbeemd, H. ed. Chemometrics Methods in Molecular Design. VCH, New York, 2: 309-318. https://doi.org/10.1002/9783527615452.ch5
[25] Efron, B., Tibshirani, R.J. (1993). An Introduction to the Bootstrap. Chapman & Hall.
[26] Wehrens, R., Putter, H., Buydens, L.M.C. (2000). The bootstrap: A tutorial. Chemometrics and intelligent Laboratory Systems, 54(1): 35-52. https://doi.org/10.1016/S0169-7439(00)00102-7
[27] Shi, L.M., Fang, H., Tong, W., Wu, J., Perkins, R., Blair, R.M., Branham, W.S., Dial, S.L., Moland, C.L., Sheehan, D.M. (2001). QSAR models using a large diverse set of estrogens. Journal of Chemical Information and Computer Sciences, 41(1): 186-195. https://doi.org/10.1021/ci000066d
[28] Hansen, O.C. (2004). Quantitative structure-activity relationships (qsar) and pesticides. Teknologisk institute. Pesticides Research no. 94.
[29] Golbraikh, A., Tropsha, A. (2002). Beware of q2. Journal of Molecular Graphics and Modelling, 20(4): 269-276. https://doi.org/10.1016/S1093-3263(01)00123-1
[30] Todeschini, R., Consonni, V., Mannhold, R. (2000). Handbook of Molecular Descriptors. Mannhold, R., Kubinyi, H., Timmerman, H. eds. Wiley- VCH Verlag G m b H. Weinheim.
[31] Ramsay, L.F., Schafer, W.D. (1997). The statistical sleuth, Belmont: Wadsworth Publishing Company.
[32] Holder, A.J., Yourtee, D.M., White, D.A., Galaros, A.G., Smith, R.J. (2003). Chain melting temperature estimation for phosphatidyl cholines by quantum mechanically derived quantitative structure property relationships. Journal of Computer-Aided Molecular Design, 17(2-4): 223-230. https://doi.org/10.1023/A:1025382226037
[33] Wang, W.J., Xu, Z.B., Lu, W.Z., Zhang, X.Y. (2003). Determination of the spread parameter in the Gaussian kernel for classification and regression. Neurocomputing, 55(3-4): 643-663. https://doi.org/10.1016/S0925-2312(02)00632-X
[34] Consonni, V., Todeschini, R., Pavan, M. (2002). Structure/response correlations and similarity/diversity analysis by GETAWAY descriptors. 1. Theory of the novel 3D molecular descriptors. Journal of Chemical Information and Computer Sciences, 42(3): 682-692. https://doi.org/10.1021/ci015504a
[35] Consonni, V., Todeschini, R., Pavan, M., Gramatica, P. (2002). Structure/response correlations and similarity/diversity analysis by GETAWAY descriptors. 2. Application of the novel 3D molecular descriptors to QSAR/QSPR studies. Journal of Chemical Information and Computer Sciences, 42(3): 693-705. https://doi.org/10.1021/ci0155053
[36] Hansch, C., Leo, A. (1979). Substituent Constants for Correlation Analysis in Chemistry and Biology. Wiley. Interscience, New York.
[37] Estrada, E. (1996). Spectral moments of the edge adjacency matrix in molecular graphs. 1. Definition and applications to the prediction of physical properties of alkanes. Journal of Chemical Information and Computer Sciences, 36(4): 844-849. https://doi.org/10.1021/ci950187r
[38] Estrada, E. (1998). Spectral moments of the edge adjacency matrix in molecular graphs. 3. Molecules containing cycles. Journal of Chemical Information and Computer Sciences, 38(1): 23-27. https://doi.org/10.1021/ci970030u