Hussein Neama Najeeb*![]() | Khalid Mahdi Jasim
| Khalid Mahdi Jasim![]() | Mohammed Ridha Shaeed Janabi
| Mohammed Ridha Shaeed Janabi![]() | Zahraa Fahad Abd Al-Sada
| Zahraa Fahad Abd Al-Sada![]() | Ghada Ali Mijbel
| Ghada Ali Mijbel![]() | Sajjad Abbas Hadi Nukhailawi
| Sajjad Abbas Hadi Nukhailawi![]()
© 2026 The authors. This article is published by IIETA and is licensed under the CC BY 4.0 license (http://creativecommons.org/licenses/by/4.0/).
OPEN ACCESS
This study aims to examine the electronic and thermoelectric properties of sodium 4-[(4-dimethylamino) phenylazo] benzene sulfonate (SE), an organic semiconducting compound with promising applications. While previous research has focused primarily on the optical behavior of this molecule, the impact of structural modifications, particularly the incorporation of amino groups on its thermoelectric performance, remains underexplored. Density Functional Theory (DFT) and Time-Dependent DFT (TD-DFT) methods were utilized to analyze five SE derivatives differing in the number of amino substituents. The results indicated that elevation of the number of amino groups would enhance charge transport, raise the Seebeck coefficient, and improve thermoelectric efficiency. Notably, the SE-5 derivative demonstrated the highest performance indicators. These enhancements are attributed to molecular structure variations and electronic interference effects, suggesting that SE derivatives hold potential for future thermoelectric and nano electronic applications.
Density Functional Theory, Sodium 4-[(4-dimethylamino) phenylazo] benzene sulfonate, modulating, electronic, strength
The rising demand for flexible and efficient energy conversion systems has led to growing interest in organic thermoelectric materials. These materials offer advantages, including low-cost synthesis, structural tunability, and mechanical flexibility, making them suitable for advanced energy-harvesting technologies. Among various classes of organic semiconductors, molecules with conjugated systems and donor–acceptor characteristics have presented promising thermoelectric behavior through controlled manipulation of their frontier molecular orbitals and charge transport properties [1].
Sodium 4-[(4-dimethylamino) phenylazo] benzene sulfonate (SE) is one organic semiconductor chemical belonging to the class of nonpolymeric semiconducting materials. It is characterized by its unusual capacity to change hue in response to pH variations and typically manifests as orange powdery crystals [2]. The electrical response of sodium 4-[(4-dimethylamino) phenylazo] benzene sulfonate to several outside factors was explored in an investigation by De Oliveira and colleagues [3]. They focused on how these stimulus types cause changes in the dye's molecular structure. SE molecules were incorporated into a solid film matrix as part of the study, allowing controlled testing as well as observation of the material's characteristics under different circumstances.
The process by which the energy of light promotes an alteration in the atoms' arrangement within a molecule, leading to different structural configurations or isomers, is known as photo-isomerization. This characteristic is particularly useful in fields where materials with the ability to change how they react in reaction to light are highly desired [4, 5]. Numerous studies have indicated how thoroughly the spectroscopic and optical characteristics of SE molecules have been examined in the literature [6-8]. These studies elucidated the emission properties of SE molecules as well as their electronic transitions along with absorption spectra. Nevertheless, there is still a great deal to learn about their thermoelectric and electrical characteristics, in spite of the abundance of information available for their optical activity. Since they can transform temperature variations into electrical voltage or vice versa, thermoelectric materials are critical for converting energy applications. This work seeks to close this gap through examining the impact of both the position and the number of groups of amines on the thermoelectric properties of SE-based molecular junctions.
This study attempts to clarify the complex interactions between molecular structure and quantum mechanical phenomena that control the performance of SE molecules in energy conversion applications, while also addressing current knowledge gaps about their thermoelectric features.
This study utilized the Gaussian09 software package, in conjunction with the IESTA programs, to perform gas-phase molecular simulations. Gaussian09 is particularly well-known for its capacity to perform quantum mechanical calculations on molecular assemblies, allowing it to explore electronic structures as well as predict molecular behavior with remarkable accuracy.
Gauss View 5.0.8 was employed to help create input files for Gaussian09 and effectually view molecular orbital structures. This graphical user interface ameliorates user interaction with Gaussian09 by offering simple tools for generating molecules in addition to ascertaining computational outcomes.
The hybrid functional B3LYP was utilized owing to its well-established performance in modeling the electronic structure of organic and organometallic systems. The B3LYP approach, which integrates Becke’s three-parameter exchange functional with the Lee–Yang–Parr correlation functional, provides a good balance between computational efficiency and accuracy, especially in systems where electron delocalization and π-conjugation play a significant role [9, 10].
In order to accurately describe the molecular system containing both light elements and heavy atoms such as gold, a dual-basis strategy was adopted. The LANL2DZ basis set was applied to gold atoms. This basis set includes effective core potentials (ECPs) which simplify the treatment of inner-core electrons and account for relativistic effects, which are non-negligible for heavy transition metals [11, 12].
For the lighter atoms (C, H, N, O, and S), the 6-31G(d) basis set was selected. This split-valence basis with polarization functions has been widely validated for organic molecules and is appropriate for describing electronic polarization, lone-pair interactions, as well as geometrical flexibility in conjugated systems [13].
This computational framework has been successfully adopted in previous studies addressing charge transport in molecular junctions involving gold electrodes and thiol-terminated organic molecules [14, 15]. Thus, the selected combination of functional and basis sets is well-suited for capturing the electronic features essential to the quantum interference and charge transport behavior examined in this work.
The study's findings revealed that the transmission coefficient T(E) is heavily affected by the electronic configuration of SE, namely the number and placement of amine groups. The calculated T(E) values demonstrate that specific topologies for amine substitution can result in better electron transport properties, which enhance thermoelectric performance. The goal of the study was to determine how the quantity and placement of amino groups would impact the molecular characteristics of the following five compounds of the SE chemical-based:
1. SE-1, with no amino groups, acts as a baseline in this study. This absence enables us to appraise the basic properties of the base structure.
2. SE-2 adds a single amino group to its structure. The presence of this group starts to alter the solubility and electrical characteristics, revealing how even a single substitution can affect molecular behavior.
3. SE-3 has two amino groups, which increases the possibility of hydrogen bonding and modifies the molecule's steric properties.
4. SE-4 contains three amino groups, which considerably raises the possibilities for intermolecular interactions and may affect its stability as well as solubility when mixed with different solvents.
5. SE-5, with four amino groups, is a highly functionalized version with potentially unique redox characteristics caused by its augmented electron-donating ability.
According to Figure 1, incorporating heteroatoms such as oxygen, nitrogen, and sulfur boosts their utility as redox indicators compounds that can experience oxidation or reduction reactions and exhibit modifications in the chemical environment. Exploration of these molecules provides valuable insights into how structural modifications can tailor their chemical properties [9].

Figure 1. Sodium 4-[(4-dimethylamino) phenylazo] benzene sulfonate (SE) compounds
The data presented in Figure 1 and Table 1 indicate a significant trend in the molecular structure of the compounds under investigation, specifically highlighting an increase in the number of amino groups from zero in molecule SE-1 to four in molecule SE-5. The amino group, characterized by its functional group formula (-NH2), is recognized for its role as a radical species owing to the presence of an unpaired electron. This unique electronic configuration contributes to the high reactivity of amino groups, making them critical in various chemical reactions and biological processes [16, 17].
Table 1. The number of amino groups (N), molecular length (l), theoretical electrode separation (Z = dAu..Au-0.25), and electrode snapback at 0.25 nm
|
Molecule |
N |
l (nm) |
d (nm) |
Z (nm) |
|
SE-1 |
0 |
1.55 |
2.116 |
1.866 |
|
SE-2 |
1 |
1.51 |
2.148 |
1.898 |
|
SE-3 |
2 |
1.48 |
2.21 |
1.96 |
|
SE-4 |
3 |
1.46 |
2.206 |
1.956 |
|
SE-5 |
4 |
1.44 |
2.2 |
1.95 |
The entire length (l) of the molecule is affected by this torsional strain, as observed in Table 1. For instance, SE-1, the longest molecule analyzed, measured 1.55 nm in length. In contrast, the molecule SE-5 is shorter, measuring 1.44 nm. The lengths of the molecules SE-2, SE-3, and SE-4 are 1.51 nm, 1.48 nm, and 1.46 nm, respectively. These variations in molecular length are caused by the altered numbers of amino groups in each molecule as well as their belongings on steric hindrance and electronic interactions within the molecular structure [18, 19].
The presence of amino groups (–NH2) in organic molecules significantly enhances their reactivity owing to the nucleophilic nature of the nitrogen atom. This elevated reactivity is not the only consequence of incorporating amino groups; it also introduces a new parameter linked to molecular conformation, specifically the twisting or torsional strain within the molecule [20]. As depicted in Figure 2, the introduction of additional amino groups contributes to a rise in inherent strain, subsequently leading to greater twisting of the molecular structure.
The observed outcomes can be explained by the twisting of the molecular structure, which occurs as the amino groups number grows from 0 to 4. This is attributed to the structural as well as electronic effects that arise from the addition of these functional groups, leading to alterations in the conformation of the molecule. In order to construct a theoretical model of the molecular junctions, 8 layers of (111)-oriented bulk gold were utilized, with individually layer comprising six atoms. 0.235 nm was kept as the interlayer spacing, which is in accordance with the usual metallic bonding in gold.
These layers were continuously repeated to generate gold electrodes that could maintain a constant current flow, approximating the perfect conditions for electrical conduction. Two pyramidal (111)-oriented gold tips were incorporated into each electrode's design and attached to the molecular structure under study. This arrangement facilitates electron transfer processes through permitting the electrodes as well as the molecule to have the best possible contact. It should also be noted that within this conceptual framework, once the atoms in each electrode have made contact with the molecule, they are allowed to relax even more. Any possible strain or distortions brought on by interactions at the interface between the electrodes as well as the molecular junction are taken into account by this relaxation process.
Methyl molecules are essential anchor groups that aid molecules adhere to gold electrodes. Effective electronic coupling requires a close engagement, as evidenced by the 2.56 Å measurement between the methyl-contacted groups as well as the apex of each of the pyramid-shaped model gold electrodes. Density Functional Theory (DFT) techniques were employed to comprehend the structural characteristics along with the interactions of these methyl molecules with gold electrodes. In particular, the B3LYP level of theory was applied for the main optimization of gas-phase molecules [21, 22].
This optimization technique made use of the 6-31G** basis set, which is ideal for researching organic compounds and has polarization functions on heavy atoms. The SIESTA (Spanish Initiative for Electronic Simulations with Thousands of Atoms) technique, which employs localized basis sets to enable effective computations on huge systems, was utilized for additional inspection. In order to guarantee a precise depiction of their interactions, every possible combination of gold with molecular orbitals was geometrically optimized in this context. For advancing the accuracy of the computations, a double-ζ polarized (DZP) basis set was selected, with a real-space grid produced with a 250 Ry energy threshold.
This choice reestablishes a balance between computational efficiency as well as accuracy in capturing electronic properties [23, 24]. The generalized gradient approximation (GGA) was employed to describe the exchange and correlation function within DFT calculations. This method accounts for non-local effects in electron density distributions, making it suitable for systems involving metallic surfaces such as gold [25, 26]. In order to ensure that all configurations reached a state of stability, geometry optimization was performed for each molecule structure under analysis until forces acting on atoms were reduced to less than 20 meV/Å. Figure 3, displaying these processes, provides visual representations of both the molecular structures involved and their interactions with gold electrodes, highlighting the significance of methyl anchoring in facilitating electronic connections. The electronic characteristics of each molecule in the gas phase were scrutinized using DFT-based techniques to gain a deeper understanding of their transport behavior. This analysis was undertaken at the B3LYP level of theory, employing the 6-31G** basis set, which is widely recognized for its balance between computational efficiency as well as accuracy in predicting molecular properties.

Figure 2. The relax structure of molecules

Figure 3. Theoretical model for molecular junction optimization
This study has a special focus on the lowest unoccupied molecular orbitals (LUMO) and the highest occupied molecular orbitals (HOMO). All of the molecules under investigation had HOMOs primarily located over their respective backbones, indicating the distinctive π−π interaction pattern that is characteristic of conjugated systems. Strong electron delocalization inside the molecular framework is demonstrated by this localization, which is indispensable for comprehending charge transport events [27, 28].
Alternatively, the sodium atoms found in the molecular structures were the main location of the LUMOs. This spatial gap between HOMO and LUMO suggests that interactions involving these atoms of sodium may have a major impact on electron transport pathways [29, 30]. An orbital analysis was performed to examine the existence, in addition to the significance of charge transfer interactions, or QI, within the orbital structures of the molecules under study to clarify this aspect further. The results of this examination indicated that, for all molecular junctions examined, charge transfer interactions are primarily responsible for controlling the transport characteristics. It was clear from Figure 4 that electron flow through these junctions is heavily affected by Collective Charge Transfer Interactions (CQI). Lambert et al. [30] have established an orbital symmetry rule [31]. This principle serves as a cornerstone for the magic ratio theory, which is predicated on the usage of the exact core of Green’s function, defined as follows [32]:
$g(E)=(I E-H)^{-1}$ (1)
One of the several approximations to $g(E)$ explored in the literature is the strategy of solely considering the contributions from the HOMO and LUMO to $\mathrm{g}(\mathrm{E})$. Since the HOMO amplitudes on sites a and b are $\psi_{\mathrm{a}}^{\mathrm{E}_{\mathrm{H}}}$ and $\psi_{\mathrm{b}}^{\mathrm{E}_{\mathrm{H}}}$, and the LUMO amplitudes on the same sites are $\psi_{\mathrm{a}}^{\mathrm{E}_{\mathrm{L}}}$ and $\psi_{\mathrm{b}}^{\mathrm{E}_{\mathrm{L}}}$, the Green's function $\mathrm{gba}(\mathrm{E})$ can be roughly represented as depicted in Eq. (2) if the inputs from the remaining orbitals are ignored:
$\mathrm{g}_{\mathrm{ab}}(\mathrm{E}) \approx \frac{\psi_{\mathrm{a}}^{\left(\mathrm{E}_{\mathrm{H}}\right)} \psi_{\mathrm{b}}^{\left(\mathrm{E}_{\mathrm{H}}\right)}}{\mathrm{E}-\mathrm{E}_{\mathrm{H}}}+\frac{\psi_{\mathrm{a}}^{\left(\mathrm{E}_{\mathrm{L}}\right)} \psi_{\mathrm{b}}^{\left(\mathrm{E}_{\mathrm{L}}\right)}}{\mathrm{E}-\mathrm{E}_{\mathrm{L}}}$ (2)
where, EH is the energy of the HOMO, and EL stands for the energy of the LUMO.
In scenarios where the product of the (HOMO, denoted as $\psi_{\mathrm{b}}^{\left(\mathrm{E}_{\mathrm{H}}\right)} \psi_{\mathrm{a}}^{\left(\mathrm{E}_{\mathrm{H}}\right)}$, and the LUMO, represented as $\psi_{\mathrm{b}}^{\left(\mathrm{E}_{\mathrm{L}}\right)} \psi_{\mathrm{a}}^{\left(\mathrm{E}_{\mathrm{L}}\right)}$, reveals a similar sign to that of the product of the LUMO, it is anticipated that at a certain energy level E within the range EH $\leq \mathrm{E} \leq \mathrm{EL}$ (which corresponds to an energy level situated within the HOMO-LUMO gap), the right side of Eq. (2) will vanish. This can be interpreted as a case of destructive interference between the HOMO and LUMO contributions.
In contrast, if the signs of these products are opposite, then the right side of Eq. (2) will persist within the HOMO-LUMO gap, suggesting constructive interference between the HOMO and LUMO. Note that destructive interference may still occur at other energy levels E outside this gap. Further, Eq. (2) may provide a limited estimate, since contributions from all other orbitals significantly influence gba(E) once the right side of the equation disappears. According to the Coulson–Rushbrooke (CR) theorem, if both indices a and b are either even or odd, then the orbital products on opposing sides of Eqs. (3) and (4) will share identical signs.
$\Psi_{\mathrm{a}}^{\left(\mathrm{E}_{\mathrm{n}}\right)} \psi_{\mathrm{b}}^{\left(\mathrm{E}_{\mathrm{n}}\right)}=\psi_{\mathrm{a}}^{\left(-\mathrm{E}_{\mathrm{n}}\right)} \psi_{\mathrm{b}}^{\left(-\mathrm{E}_{\mathrm{n}}\right)}$ (3)
$\phi_{\mathrm{b}}^{\left(\mathrm{E}_{\mathrm{n}}\right)} \phi_{\mathrm{b}}^{\left(\mathrm{E}_{\mathrm{n}}\right)}=\phi_{\mathrm{b}}^{\left(-\mathrm{E}_{\mathrm{n}}\right)} \phi_{\mathrm{b}}^{\left(-\mathrm{E}_{\mathrm{n}}\right)}$ (4)
The eigenstate corresponding to the energy level of -En is intrinsically linked to the eigenstate associated with the energy level of En. Notably, the eigenvalues ±En manifest in pairs, demonstrating a symmetrical relationship. While Eq. (2) presents efficacy in describing bipartite lattices, it can be reasonably posited that this equation serves as a reliable approximation for other types of lattices as well. Nevertheless, it is essential to recognize that the phenomenon of exact cancellation is characteristic solely of bipartite lattices. As highlighted by Yoshizawa and his group in their studies [33], the interference patterns between the HOMO and the LUMO—whether constructive or destructive—can be effectively analyzed through examining orbital colors. This analysis is facilitated by the fact that orbital representations, such as those depicted in Figure 2, are often readily obtainable from DFT computations. To streamline our discussion further, we can reformulate Eq. (2) as follows:
$\mathrm{g}_{\mathrm{ab}}(\mathrm{E}) \approx \frac{\mathrm{a}_{\mathrm{H}}}{\mathrm{E}-\mathrm{E}_{\mathrm{H}}}+\frac{\mathrm{a}_{\mathrm{L}}}{\mathrm{E}-\mathrm{E}_{\mathrm{L}}}$ (5)
where, $a_H=\psi_a^{\left(E_H\right)} \psi_b^{\left(E_H\right)}$ and $a_L=\psi_a^{\left(E_L\right)} \psi_b^{\left(E_L\right)}$.
For certain energy values E within the range EH ≤ E ≤ EL, the right side of Eq. (5) will become null if the products of the HOMO aH and the LUMO aL share the same sign. In other words, at specific energies located within the HOMO-LUMO gap, destructive interference occurs between these orbitals. Nevertheless, it should be noted that this does not necessarily mean that the precise function gba(E) will vanish entirely. Indeed, when the right-hand side of Eq. (5) approaches zero, contributions from other orbitals may dominate over these leading terms. This observation holds promise for characterizing qualitative properties of molecules as well as for detecting quantum interference effects [34, 35].
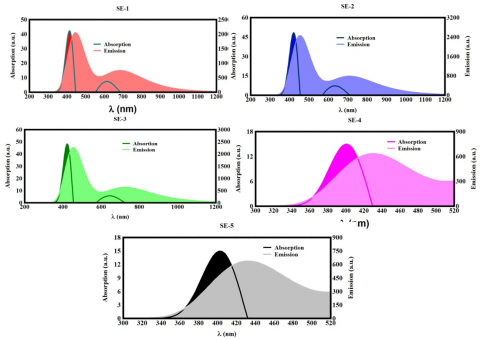
Numerous research studies have increasingly focused on the unique characteristics of SE molecules, especially their spectroscopic properties, which include both absorption and emission spectra. These spectroscopic characteristics are critical for understanding the behavior and applications of these compounds in various fields such as materials science, photonics, and biochemistry. In particular, Table 2 reveals that these SE compounds have emission spectra between 430 and 456 nm as well as absorption spectra between 399.2 and 420.8 nm. Molecular strains as along with conformational twisting brought on by the presence of amino groups within the molecular structure, are two possible explanations for the discovered asymmetry in these spectrum characteristics. It has been indicated that the intrinsic strain within the molecule grows with the number of amino groups. Significant twisting of the molecular structure could emanate from this strain, changing the structure and also spectroscopic characteristics [36, 37].

Figure 4. The red portion is a negative sign, while the blue portion is a positive sign, reflecting the optimized shape of every molecule in a gas phase. The product of the highest occupied molecular orbitals and lowest unoccupied molecular orbitals amplitudes is aH.aL
Table 2. N, highest occupied molecular orbitals, lowest unoccupied molecular orbitals, H-L gap, AλMax, EλMax, fem, and SS values for the molecules under study
|
Molecule |
N |
HOMO (eV) |
LUMO (eV) |
H-L gap (eV) |
AλMax (nm) |
EλMax (nm) |
Fem |
SS (nm) |
|
SE-1 |
0 |
4.14 |
1.7 |
2.44 |
413.2 |
446.8 |
0.0465 |
33.6 |
|
SE-2 |
1 |
4.18 |
1.77 |
2.41 |
420.8 |
452.8 |
0.0564 |
32 |
|
SE-3 |
2 |
3.9 |
1.52 |
2.38 |
424 |
456 |
0.0558 |
32 |
|
SE-4 |
3 |
4.07 |
1.72 |
2.35 |
399.2 |
430 |
0.0355 |
30.8 |
|
SE-5 |
4 |
3.92 |
1.60 |
2.32 |
401.6 |
430.4 |
0.0392 |
28.8 |
Figure 5. The UV/Vis emission and absorption spectra of every molecule
Understanding the link between twisting effects along molecular strain is essential to comprehending how these features support the reactivity and spectroscopic characteristics of SE molecules. For instance, it is notable that amino groups are completely lacking in other forms, such as SE-1. When compared to other derivatives with differing amounts of amino groups, this lack of amino groups points to a different process, considering its spectral behavior as well as structural integrity [38-41]. The results presented in reference [36] align with the findings of this study. Further, as presented in Table 2 and Figure 5, the Stokes shift values for these compounds are observed to be below 50 nm, specifically ranging from 28.8 nm to 33.6 nm. These observations suggest that SE molecules may not be appropriate for applications in medicinal fields and cryptography.
For enhancing the clarity and depth of understanding regarding electronic transitions and the impact of varying quantities of NH3 on electron transport, this study employed the Mulliken population analysis as a method to characterize electronic charge distribution. Mulliken population analysis was applied to qualitatively evaluate the charge redistribution between the molecule and the gold electrodes upon the formation of the molecular junction. This method provides a basic yet informative picture of electron transfer directionality as well as the localization of charge across the system.
Mulliken analysis is inherently sensitive to the choice of basis set. To address this limitation, all population analyses were carried out using a consistent combination of basis sets—6-31G(d) for light atoms (C, H, N, O, S) and LANL2DZ for gold atoms—throughout the entire system. This approach ensures internal consistency and minimizes artificial deviations in charge estimations owing to basis set variation [42, 43].
While absolute charge values from Mulliken analysis can be basis-set dependent and should not be interpreted quantitatively, the observed relative trends as well as changes in charge distribution, are considered reliable for drawing qualitative conclusions. To support the findings, complementary analysis such as molecular orbital visualization and electron density difference maps were also undertaken, reinforcing the direction and extent of charge transfer identified by Mulliken analysis [44]. This approach facilitates inspection of the number of electrons transferred from the molecule to the electrodes, as demonstrated in Figure 6 and detailed in Table 3. A noteworthy finding outlined in Figure 6 is that the SE-1 molecule is capable of transmitting approximately 1.1 electrons to the electrodes, whereas the SE-5 molecule demonstrates a significantly higher transmission of about 3.1 electrons.

Figure 6. Transferred electrons number from molecule to the electrodes (Γ) for all molecules
Table 3. The values of Γ, T (E), HOMO in a junction (JHOMO); LUMO in a junction (JLUMO), and JH-L gap of the molecules in a junction
|
Molecule |
Γ |
T(E) |
JHOMO (eV) |
JLUMO (eV) |
JH-L gap (eV) |
|
SE-1 |
1.1 |
4.51 × 10-5 |
0.78 |
1.58 |
2.36 |
|
SE-2 |
1.5 |
4.67 × 10-5 |
0.72 |
1.62 |
2.34 |
|
SE-3 |
2.3 |
6.76 × 10-5 |
0.59 |
1.96 |
2.55 |
|
SE-4 |
2.7 |
7.22 × 10-5 |
0.53 |
2.01 |
2.54 |
|
SE-5 |
3.1 |
7.59 × 10-5 |
0.46 |
2.48 |
2.94 |
The values of the Seebeck coefficient (S) and the dimensionless figure of merit (ZelT) are derived from the slope of the temperature-dependent energy (T(E)) relationship. The power factor (P), together with S and ZelT, can be calculated by the following equations:
$\mathrm{S} \approx-\mathrm{L}|\mathrm{e}| \mathrm{T}\left(\frac{\mathrm{d} \ln \mathrm{T}(\mathrm{E})}{\mathrm{dE}}\right)_{\mathrm{E}=\mathrm{E}_{\mathrm{F}}}$ (6)
The Lorenz number, denoted as L, is defined by the equation $\mathrm{L}=\left(\frac{\mathrm{k}_{\mathrm{B}}}{\mathrm{e}}\right)^2 \frac{\pi^2}{3}=2.44 \times 10^{-8}$. This relationship reveals that the Seebeck coefficient, S, is directly proportional to the negative of the slope of lnT(E) when ascertained at the Fermi energy level. Utilizing the Seebeck coefficient as a foundational parameter, the power factor can subsequently be calculated.
$P=G S^2 T$ (7)
where, T = 300 K represents temperature, G and S denote electrical conductance and Seebeck coefficient; ZelT, which is the purely electronic figure of merit, is given as [45]:
$\mathrm{Z}_{\mathrm{el}} \mathrm{T}=\frac{\mathrm{S}^2 \mathrm{G}}{\mathrm{k}_{\mathrm{el}}} \mathrm{T}=\frac{\mathrm{S}^2}{\mathrm{~L}}$ (8)
where, kel = electron thermal conductance. The ZelT for this research has only been determined based purely on electronic contribution, in accordance with other studies [46-48]. The results exhibited in Figure 7 reflect several facts: First, the value of (S) is very sensitive to the position of the Fermi energy, as it is noted that it changes dramatically whenever the position of the Fermi energy changes. Further, at the theoretical Fermi energy (0.0 eV), the highest Seebeck coefficient value (36.9 μVK-1) is provided by the molecule SE-5. In contrast, the lowest value for the Seebeck coefficient (16.8 μVK-1) was found for molecule SE-1, as outlined in Table 4. These results may be attributed to the presence and number of the amino group, as the SE-5 molecule contains four amino groups, which in turn resulted in structural changes that affected the slope of the transmission coefficient curve, giving the highest value for the Seebeck coefficient [49, 50]. In this context, the order of the Seebeck coefficient is SSE-5 > SSE-4 > SSE-3 > SSE-2 > SSE-1. Finally, the sign of the Seebeck coefficient for all molecules is positive. This reflects the fact that the transport process is dominated by HOMO orbitals. The efficient conversion of an input heat to electricity is a well-known characteristic of the performance of thermoelectric materials [51, 52].
It is crucial to improve the electronic figure of merit (ZelT) and power factor (P), which rely on the Seebeck coefficient (S). According to Table 4 and Figure 8, molecule SE-5 presented the highest ZelT values (0.05), while molecule SE-1 indicated the lowest values (0.011). The ZelT was also found in the following order: ZelT SE-5 > ZelT SE-4 > ZelT SE-3 > ZelT SE-2 > ZelT SE-1. These findings not only revealed how the presence and quantity of amino groups improve S and ZelT, but also showed how the inherent strain in SE molecules plays a key role in altering the slope of T(E), which enhances the molecules' thermoelectric properties. Further, the power factor sequence was PSE-5 > PSE-4 > PSE-3 > PSE-2 > PSE-1 owing to the conflict between electrical conductance and the Seebeck coefficient as determined by Eq. (6). These compounds may be viable options for thermoelectric applications in view of the previously described findings.

Figure 7. Electronic figure of merit (ZelT) as a function of the Fermi energy
Table 4. Electrical conductance (G); Seebeck coefficient (S); Electronic figure of merit (ZelT); Power factor (P) for all molecular junctions
|
Molecule |
S μVK-1 |
P WK-1 × 10-20 |
ZelT |
|
SE-1 |
16.8 |
149.58 |
0.011 |
|
SE-2 |
20 |
248 |
0.015 |
|
SE-3 |
26.6 |
558.97 |
0.027 |
|
SE-4 |
29.6 |
771.02 |
0.032 |
|
SE-5 |
36.9 |
1307.14 |
0.05 |

Figure 8. Seebeck coefficient (S) as a function of the Fermi energy
The current-voltage (I-V) properties of all molecular junctions, which are restricted to the 1st and 3rd quadrants of the I-V plane spanning the origin, are depicted in Table 4 and Figure 9. As such, they are categorized as components that use electricity, which is where the threshold voltage (Vth) value finds significance. These molecules are attractive candidates for electrical applications since their Vth values range from 0.4 to 0.7 V. All molecular junctions' I-V characteristics also revealed a quantum staircase structure in the conductance. Needless to say, when the voltage rises, the electron density grows as well, elevating the number of occupied sub-bands. In this instance, dependence conductance is a collection of plateaus divided by height 2e2/h steps: each time the Fermi level coincides with one of the sub-bands, the conductance of molecular channels changes sequentially. Thus, the adiabatic transparency of spin-nondegenerate subbands of these molecules may be responsible for the quantum staircase behavior [8, 53, 54].

Figure 9. The studied molecular junctions and their current-voltage characteristics
This study examined the influence of amino group substitution on the structural and electronic properties of SE molecules, with an emphasis on their potential roles in thermoelectric and molecular electronic applications. The findings indicated that the presence of amino groups induces significant intrinsic strain, giving rise to molecular twisting and a reduction in the overall molecular length. These structural modifications, combined with the constructive quantum interference (CQI) effect, lead to an enhancement of the transmission function T(E), contributing to improved electronic behavior.
The computed high Seebeck coefficient (S) values strongly support the potential of these molecules for thermoelectric applications. Further, the observed low threshold voltages, along with distinct semiconductor behavior and the manifestation of a quantum staircase in the transmission spectrum, suggest that these molecules are promising for various organic electronic devices.
Nevertheless, the study was primarily theoretical, relying on DFT and TD-DFT calculations. As such, experimental validation remains a crucial next step. Factors such as environmental stability, electrode interactions, and practical device performance were not examined and represent limitations of the current work.
Future research should focus on experimental synthesis and characterization of the proposed structures, the exploration of alternative functional group substitutions, and the integration of these molecules into real devices to ascertain their practical performance in energy conversion and nano electronics.
[1] Zhang, Y., Park, S.J. (2019). Flexible organic thermoelectric materials and devices for wearable green energy harvesting. Polymers, 11(5): 909. https://doi.org/10.3390/polym11050909
[2] Stojković, D., Ivanović, M., Ilić, V. (2012). Ionization of methyl orange in aqueous sodium chloride solutions. The Journal of Chemical Thermodynamics, 53: 93-99. https://doi.org/10.1016/j.jct.2012.04.023
[3] de Oliveira, H.P., Oliveira, E.G.L., de Melo, C.P. (2006). Aggregation of methyl orange probed by electrical impedance spectroscopy. Journal of Colloid and Interface Science, 303(2): 444-449. https://doi.org/10.1016/j.jcis.2006.08.019
[4] Dhammika Bandara, H.M.D., Burdette, S.C. (2012). Photoisomerization in azobenzene and related systems. Chemical Society Reviews, 41(5): 1809-1825. https://doi.org/10.1039/C2CS35170E
[5] Natansohn, A., Rochon, P. (2002). Photoinduced motions in azo-containing polymers. Chemical Reviews, 102(11): 4139-4176. https://doi.org/10.1021/cr970155y
[6] Fang, Y., Li, H., Zhang, X. (2025). Thermoelectric and thermal properties of molecular junctions: Mechanisms, characterization methods and applications. Chemical Communications, 61: 4447-4464. https://doi.org/10.1039/D4CC06822J
[7] Gonzalez‑Casal, F., Jouclas, R., Arbouch, I., Geerts, Y.H., van Dyck, C., Cornil, J., Vuillaume, D. (2024). Thermoelectric properties of benzothieno‑benzothiophene self‑assembled monolayers in molecular junctions. The Journal of Physical Chemistry Letters, 15(46): 11593-11600. https://doi.org/10.1021/acs.jpclett.4c02753
[8] Al‑Mohana, S.M.S., Najeeb, H.N., Al‑Utayjawee, R.M., Babaei, F., Al‑Owaedi, O.A. (2024). Theoretical investigation of thermoelectric properties of methyl blue‑based molecular junctions. RSC Advances, 14(33): 23699-23709. https://doi.org/10.1039/D4RA03574G
[9] Becke, A.D. (1993). Density-functional thermochemistry. III. The role of exact exchange. Journal of Chemical Physics, 98(7): 5648-5652. https://doi.org/10.1063/1.464913
[10] Lee, C., Yang, W., Parr, R.G. (1988). Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Physical Review B, 37: 785-789. https://doi.org/10.1103/PhysRevB.37.785
[11] Hay, P.J., Wadt, W.R. (1985). Ab initio effective core potentials for molecular calculations. Journal of Chemical Physics, 82(1): 270-283. https://doi.org/10.1063/1.448799
[12] Roy, L.E., Hay, P.J., Martin, R.L. (2008). Revised basis sets for the LANL effective core potentials. Journal of Chemical Theory and Computation, 4(7): 1029-1031. https://doi.org/10.1021/ct8000409
[13] Hehre, W.J., Ditchfield, R., Pople, J.A. (1972). Self-consistent molecular orbital methods. XII. Further extensions of gaussian—type basis sets for use in molecular orbital studies of organic molecules. Journal of Chemical Physics, 56(5): 2257-2261. https://doi.org/10.1063/1.1677527
[14] Solomon, G.C., Andrews, D.Q., Hansen, T., Goldsmith, R.H., Wasielewski, M.R., Van Duyne, R.P., Ratner, M.A. (2010). Understanding quantum interference in coherent molecular conduction. Journal of Chemical Physics, 129(5): 054701. https://doi.org/10.1063/1.2958275
[15] Guédon, C.M., Valkenier, H., Markussen, T., Thygesen, K.S., Hummelen, J.C., van der Molen, S.J. (2012). Observation of quantum interference in molecular charge transport. Nature Nanotechnology, 7: 305-309. https://doi.org/10.1038/nnano.2012.37
[16] Capelli, R., Toffanin, S., Generali, G., Usta, H., Facchetti, A., Muccini, M. (2010). Organic light-emitting transistors with an efficiency that outperforms the equivalent light-emitting diodes. Nature Materials, 9(6): 496-503. https://doi.org/10.1038/nmat2751
[17] Dazzo, E., Rehberg, K., Michelucci, R., Passarelli, D., Boniver, C., Vianello Dri, V., Striano, P., Striano, S., R. Pasterkamp, J., Nobile, C. (2018). Mutations in MICAL-1cause autosomal-dominant lateral temporal epilepsy. Annals of Neurology, 83(3): 483-493. https://doi.org/10.1002/ana.25167
[18] Zeise, M., Stojanović, M., Baranac‑Stojanović, M. (2015). The effect of steric repulsion on the torsional potential of n‑butane: A theoretical study. Tetrahedron, 71(32): 5119-5123. https://doi.org/10.1016/j.tet.2015.06.002
[19] Flahaut, D., Minvielle, M., Sambou, A., Lecour, P., Legens, C., Barbier, J. (2017). Identification of sulphur, oxygen and nitrogen species in heavy oils by X-ray photoelectron spectroscopy. Fuel, 202: 307-317. https://doi.org/10.1016/j.fuel.2017.04.046
[20] Foner, S.N. (1964). Free radicals and unstable molecules. Science, 143(3605): 441-450. https://doi.org/10.1126/science.143.3605.441
[21] Halboos, S.H., Al-Owaedi, O.A., Al-Robayi, E.M. (2024). Quantum interference features and thermoelectric properties of macrocyclic-single molecules: Theoretical and modelling investigation. Nanoscale Advances, 6(24): 6303-6316. https://doi.org/10.1039/d4na00541d
[22] Higuchi, J. (1956). Electronic structures of NH, NH2, and NH3. The Journal of Chemical Physics, 24(3): 535-545. https://doi.org/10.1063/1.1742542
[23] Hille, R., Hall, J., Basu, P. (2014). The mononuclear molybdenum enzymes. Chemical Reviews, 114(7): 3963-4038. https://doi.org/10.1021/cr400443z
[24] Homocianu, M., Airinei, A., Dorohoi, D.O. (2011). Solvent effects on the electronic absorption and fluorescence spectra. Journal of Advanced Research in Physics, 2(1): 1-9.
[25] Hung, N.T., Nugraha, A.R.T., Saito, R. (2019). Thermoelectric properties of carbon nanotubes. Energies, 12: 4561. https://doi.org/10.3390/en12234561
[26] Keal, T.W., Tozer, D.J. (2004). A semiempirical generalized gradient approximation exchange-correlation functional. Journal of Chemical Physics, 121(12): 5654-5660. https://doi.org/10.1063/1.1784777
[27] Kravchenko, S.V., Kravchenko, G.V., Furneaux, J.E., Pudalov, V.M., D’Iorio, M. (1994). Possible metal-insulator transition at B=0 in two dimensions. Physical Review B, 50(11): 8039-8042. https://doi.org/10.1103/PhysRevB.50.8039
[28] Krishna, P., Vijay, V., Ponnusamy, S., Navaneethan, M. (2024). Mass and strain field mediated low thermal conductivity for enhanced thermoelectric properties in Zn substituted SnS. CrystEngComm, 26(40): 5767-5776. https://doi.org/10.1039/d4ce00627e
[29] Kutzelnigg, W. (1996). Friedrich Hund und die Chemie. Angewandte Chemie, 108(6): 629-643. https://doi.org/10.1002/ange.19961080604
[30] Lambert, C.J., Liu, S.X. (2018). A magic ratio rule for beginners: A chemist’s guide to quantum interference in molecules. Chemistry - A European Journal, 24(17): 4193-4201. https://doi.org/10.1002/chem.201704488
[31] Lao, K.Q., Person, M.D., Xayariboun, P., Butler, L.J. (1990). Evolution of molecular dissociation through an electronic curve crossing: Polarized emission spectroscopy of CH3I at 266 nm. The Journal of Chemical Physics, 92(2): 823-841. https://doi.org/10.1063/1.458116
[32] Li, F., Li, T., Zhang, L., Jin, Y., Hu, C. (2021). Enhancing photocatalytic performance by direct photo-excited electron transfer from organic pollutants to low-polymerized graphitic carbon nitride with more C-NH/NH2 exposure. Applied Catalysis B: Environmental, 296: 120316. https://doi.org/10.1016/j.apcatb.2021.120316
[33] Li, P., Wang, Z., Song, C., Zhang, H. (2017). Rigid fused π-spacers in D-π-A type molecules for dye-sensitized solar cells: A computational investigation. Journal of Materials Chemistry C, 5(44): 11454-11465. https://doi.org/10.1039/c7tc03112b
[34] Majeed, H.A., Sharba, A.B. (2022). Optical characterization of Methylene blue over the whole visible range. AIP Conference Proceedings, 2547(1): 030006. https://doi.org/10.1063/5.0112497
[35] Makhova, N.N., Belen’kii, L.I., Gazieva, G.A., Dalinger, I.L., Konstantinova, L.S., Kuznetsov, V.V., Kravchenko, A.N., Krayushkin, M.M., Rakitin, O.A., Starosotnikov, A.M., Fershtat, L.L., Shevelev, S.A., Shirinian, V.Z., Yarovenko, V.N. (2020). Progress in the chemistry of nitrogen-, oxygen- and sulfur-containing heterocyclic systems. Russian Chemical Reviews, 89(1): 55-124. https://doi.org/10.1070/rcr4914
[36] Miller, J.S. (1990). Molecular Materials I. Molecular materials mimic inorganic network solids. Advanced Materials, 2(2): 98-99. https://doi.org/10.1002/adma.19900020207
[37] Ozawa, H., Baghernejad, M., Al-Owaedi, O.A., Kaliginedi, V., Nagashima, T., Ferrer, J., Wandlowski, T., García-Suárez, V.M., Broekmann, P., Lambert, C.J., Haga, M.A. (2016). Synthesis and single-molecule conductance study of redox-active ruthenium complexes with pyridyl and dihydrobenzo[b]thiophene anchoring groups. Chemistry - A European Journal, 22(36): 12732-12740. https://doi.org/10.1002/chem.201600616
[38] Pellin, M.J., Foosnaes, T., Gruen, D.M. (1981). Fluorescence spectrum of Mo2 in argon and krypton matrices. The Journal of Chemical Physics, 74(10): 5547-5557. https://doi.org/10.1063/1.440917
[39] Perdew, J.P., Chevary, J.A., Vosko, S.H., Jackson, K.A., Pederson, M.R., Singh, D.J., Fiolhais, C. (1992). Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Physical Review B, 46(11): 6671-6687. https://doi.org/10.1103/PhysRevB.46.6671
[40] Putatunda, A., Singh, D.J. (2019). Lorenz number in relation to estimates based on the Seebeck coefficient. Materials Today Physics, 8: 49-55. https://doi.org/10.1016/j.mtphys.2019.01.001
[41] Rindt, I.F.F., Offe, A.D.Y., Bowden, F.P. (1963). Physical properties of layer structures: Optical properties and photoconductivity of thin crystals of molybdenum disulphide. Proceedings of the Royal Society of London. Series A. Mathematical and Physical Sciences, 273(1352): 69-83. https://doi.org/10.1098/rspa.1963.0075
[42] Reed, A.E., Weinstock, R.B., Weinhold, F. (1985). Natural population analysis. The Journal of Chemical Physics, 83(2): 735-746. https://doi.org/10.1063/1.449486
[43] Bultinck, P., Van Alsenoy, C., Ayers, P.W., Carbó-Dorca, R. (2007). Critical analysis and extension of the Hirshfeld atoms in molecules. The Journal of Chemical Physics, 126(14): 144111. https://doi.org/10.1063/1.2715563
[44] Fonseca Guerra, C., Handgraaf, J.W., Baerends, E.J., Matthias Bickelhaupt, F. (2004). Voronoi deformation density charges: Assessment of the Mulliken, Bader, Hirshfeld, and VDD methods. Journal of Computational Chemistry, 25(2): 189-210. https://doi.org/10.1002/jcc.10351
[45] Dresselhaus, M.S., Chen, G., Tang, M.Y., Yang, R.G., Lee, H., Wang, D.Z., Ren, Z.F., Fleurial, J.P., Gogna, P. (2007). New directions for low-dimensional thermoelectric materials. Advanced Materials, 19(8): 1043-1053. https://doi.org/10.1002/adma.200600527
[46] Schnupf, U., Willett, J.L., Bosma, W.B., Momany, F.A. (2007). DFT study of α- and β-d-allopyranose at the B3LYP/6-311++G** level of theory. Carbohydrate Research, 342(2): 196-216. https://doi.org/10.1016/j.carres.2006.12.006
[47] Shakourian-Fard, M., Kamath, G., Smith, K., Xiong, H., Sankaranarayanan, S.K.R.S. (2015). Trends in Na-Ion Solvation with Alkyl-Carbonate Electrolytes for Sodium-Ion Batteries: Insights from First-Principles Calculations. Journal of Physical Chemistry C, 119(40): 22747-22759. https://doi.org/10.1021/acs.jpcc.5b04706
[48] Sharma, M., Kumari, M., Rani, S., Yadav, A.K., Solanki, P.R., Mozumdar, S. (2021). Influence of pH, β-cyclodextrin, and metal ions on the solubility and stability of the medicinally competent isoxazole derivative of curcumin: A photophysical study. ACS Applied Bio Materials, 4(12): 8407-8423. https://doi.org/10.1021/acsabm.1c00957
[49] Shelykh, I.A., Bagraev, N.T., Klyachkin, L.E. (2003). Spin Depolarization in Spontaneously Polarized Low-Dimensional Systems. Semiconductors, 37(12): 1390-1399. https://doi.org/10.1134/1.1634660
[50] Stjerna, B., Granqvist, C.G. (1990). Optical properties of SnOx thin films: Theory and experiment. Applied Physics Letters, 57(19): 1989-1991. https://doi.org/10.1063/1.104150
[51] Suwardi, A., Cao, J., Zhao, Y., Wu, J., et al. (2020). Achieving high thermoelectric quality factor toward high figure of merit in GeTe. Materials Today Physics, 14: 100239. https://doi.org/10.1016/j.mtphys.2020.100239
[52] Tian, Z., Zou, Y., Liu, G., Wang, Y., Yin, J., Ming, J., Alshareef, H.N. (2022). Electrolyte solvation structure design for sodium ion batteries. Advanced Science, 9(22): 2201207. https://doi.org/10.1002/advs.202201207
[53] Al-Owaedi, O.A., Najeeb, H.N., Obaid, A.K., Al-Daamy, M. (2024). Thermoelectric signature of d-orbitals in tripod-based molecular junctions. Materials Advances, 5(24): 9781-9791. https://doi.org/10.1039/D4MA00646A
[54] Abbas Mohammed, H., Al-Owaedi, O.A., Najeeb, H.N. (2024). Innovative applications of rotaxane-based molecular junctions in electronics and optoelectronics. Journal of Theoretical and Applied Physics, 18(4): 182455. https://doi.org/10.57647/j.jtap.2024.1804.55