OPEN ACCESS
Thermodynamic performances of Mg2Sn metal compounds were calculated by first principle calculations. This study, based on density functional theory, calculated lattice constants, band structure and elastic constants to detail the thermal stability of Mg2Sn structures, and then focused on the phonon density of states, constant volume and Debye temperature to deal with thermodynamic properties. Additionally, the calculations were compared with available experimental value and previous theoretical data, and a favorable result has been obtained in the work. Furthermore, the results manifested that Mg2Sn has better capability when adding the alloying element Sn into Mg alloys, and demonstrated that quasi-harmonic Debye model with first-principles is an efficient approach to study the behavior of intermetallic compounds, which provided more valuable instructional information for the design and application of Mg2Sn compounds.
Mg2Sn compound, Thermodynamic properties, Phonon spectrum, First principles.
Recently, Mg2X (X=Sn, Si and Ge) compounds have shown development of alloy conductor materials, which is due to its abundant raw availability and excellent thermal effect, especially its excellent thermoelectric characteristics. Such inter-A2B metal compound crystals have the features of resistance to corrosion, withstanding high temperatures, high electrical conductivity and low thermal conductivity, etc. [1]. Thereby Mg2X based thermoelectric materials have gained wide attention of researchers, and has been referred to as a new type of semiconductor material environment [2-4]. Additionally, on account of its low density and light weight, Mg2X alloy have an indispensable application value in the aerospace and automotive fields [5-7]. However, because of its lack of performance, such a magnesium alloy has been limited in the application process. Accordingly, alloying is considered to be an important means of improving the properties of magnesium alloys [8-10]. One general way of improving the physical and chemical properties of magnesium alloys is used in the application areas, which may improve the microstructure or generate precipitates by adding new alloying elements in magnesium alloys.
However, there are a series of problems with preparation Mg2X compounds, such as the large difference of the melting point between Mg and X elements, making their integration
arduous. Moreover, Mg and oxygen readily react at high temperatures, which results in a decline in the thermoelectric performance. In addition, there is a serious inter-granular brittleness, similar to other inter-metallic compounds, which leads to meaning unclear. formidably form. Therefore the deformation mechanism and microscopic mechanisms are further studied under the conditions of the large external strain and mechanical properties of material. Afterwards, it is essential to design a new magnesium alloy to understand the role of alloying elements for the stability and mechanical properties and electronic structure of Mg alloy phase.
Currently, the phase stability and mechanical and thermodynamic performance of magnesium alloy have been studied [11, 12], and literatures [13] advocate that it can effectively optimize the crystal structure and enhance the mechanical properties through adding the Sn element to magnesium alloys, and electronic structure plays a key role in semiconductor optoelectronic properties. Therefore, the calculations of band structure of A2B metal compound become a topic of great interest in the field of computational materials. In theory, Ganeshan studied the elastic properties at different temperatures [14]. Buchenauer and Tani [15,16] further calculated the electronic structure and optical properties of Mg2Sn and obtained its elastic constants and transverse optical wave frequency. Davis carried out the elastic constants and lattice dynamics of Mg2Sn and acquired the sound speed in different directions using resonance technology[17]. Moreover, Kearney reported the lattice dynamics of Mg2Sn in room temperature [18]. In addition, some results compared the experimental and theoretical values to provide more valuable instructional information for the design and application of Mg alloys [1, 19, 20].
From the above mentioned studies, existing theories have pointed to ways for researchers to obtain high-performance thermoelectric materials. Despite of the many theoretical and experimental studies of the electronic structure of Mg2Sn, those reported only referred to its dielectric function of optical properties, and this work is only in the initial stage. It is noted that the density functional method based on first principles has become an important tool to accurately calculate the solid structure, and thermal and mechanical properties. In practice, the first principles calculations depending on exact prediction and analytical methods have been widely applied in various fields of engineering [21], due to its empirical parameters which are different from other calculation methods, and further for its capacity to obtain the system energy by solving the Schrodinger equation. Additionally, its wide range of applications has been involved in estimating the material composition and structure and performance characteristics to guide the design of new materials. Moreover, it plays a supporting role in experimental results, and even to improve the simulation process when the engineering experiment cannot be achieved. Unfortunately, reports of thermodynamic properties via first principles calculations, such as phonon dispersion relations and phonon density of states and isovolumic specific heat and Debye temperature are lacking recently.
In this work, two aspects of this study must be addressed. The first examination relates to the thermal stability by investigating lattice constants and elastic constants.The second aspect deals with phonon density of states and thermodynamic parameters of Mg2Sn magnesium alloy with plane-wave pseudopotential method based on first principles, and detailing its mechanism.
First principles calculations were performed on the electronic structure using density functional theory (DFT), thermodynamic properties and phonon spectrum of Mg2Sn alloys intermetallic compounds. Initially, it must optimize the structures with full relaxation (Ultrafine, Ecut=330eV), and obtaine the lattice parameters. The semiconductor Mg2Sn has the anti-fluorite crystal structure (No.255, space group is $\mathrm{Fm}_{3}^{-} \mathrm{m}$, and the lattice constant of 0.676 nm), and its structure consists of interpenetrating the fcc cubic lattices
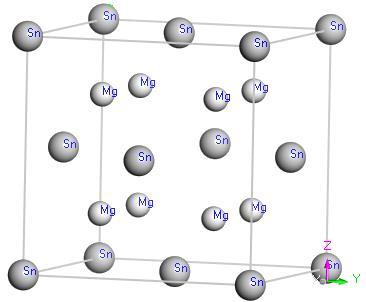
with Sn4+ at the origin (0, 0, 0) and Mg2+ at ± (1/4, 1/4, 1/4) position [22]. In addition, the coordination number of each Sn ion is 8, and each Mg-ion is located in the middle of Sn4+ tetrahedral crystal structure, as shown in Fig.1. Afterwards, in order to discuss thermodynamic properties of Mg2Sn crystals, the phonon dispersion system must firstly be calculated, and then the Mg2Sn ground state values of the lattice constant are estimated by geometrical optimization.
The related parameters were set as follows: the exchange-correlation part and ultrasoft pseudopotential between the valence electrons and atomic was described with Generalized Gradient Approximation (GGA) under the Perdew Burke and Ernzerhof (PBE) [23] gradient correction function. The Monkhorst-Pack scheme k-point sampling with 6×6×6 (unitcell cases) has been used for the integration of the Brillouin zone. When Ecut-off converged to 340eV, the lattice parameters and the atomic position are fully relaxed, and then the final forces on all relaxed atoms are less than e-005 eV·atom-1. In order to verify the validity of pseudopotential plane wave method, the study optimized the lattice constant and calculated three independent elastic constants (C11, C12, C44) according to the literature [24] after relaxing the shape and volume of unitcell, and the results listed in Table 1. The slightly higher values than the experimental values is because the use of the GGA approximation process usually overestimates lattice constants exchange-correlation function, and the visible results are well aligned with the experimental data, with reasonable calculation parameters research settings.
Figure 1. Crystal structure
Much of the research demonstrates that many properties of solid crystals are closely related to the elastic properties, such as the equation of state, specific heat capacity, Debye temperature and melting point. Some important information about the characteristics and stability of the crystal anisotropic and crystal structure on the basis of the elastic constants is obtained. Furthermore, one of the most significant methods is the use of elastic constant to calculate Debye temperature. In addition, the enthalpy of formation of Mg2Sn alloys can be calculated to determine structural stability. Subsequently, the work obtained the results of ΔH as shown in Tab.1 according to the equations [25], the calculated values are well aligned with the theoretical value, and results show that ΔH=-0.21 eV·atom-1<0,which shows that the structure of Mg2Sn alloy is more thermodynamically stable.
Table 1. Lattice constants and elastic constants of Mg2Sn phases [26-31]
|
Parameters |
Exp. |
Cal. |
|
a/Å |
6.76 |
6.82 |
|
a/Å |
6.82 |
6.82 |
|
C11 /GPa |
82.40 |
80.29 |
|
C12/GPa |
20.80 |
20.31 |
|
C44/GPa |
36.60 |
33.95 |
|
ΔH/(eV·atom-1) |
-0.26 |
-0.21 |
Figure 2. Phonon dispersion and density of phonon states
Additionally, heat capacity was obtained on the basis of the Eq. (1) and Eq. (2) [32], which helped to investigate the dependence degree of thermodynamic properties of temperature.
$C_{V}(T)=k \int \frac{(E \omega / k T)^{2} \exp (E \omega / k T)}{[\exp (E \omega / k T)-1]^{2}}$(1)
$C_{V}(T)=9 N k\left(\frac{T}{\Theta_{D}}\right)^{3} \int_{0}^{\Theta_{\rho} / T} \frac{X^{4} e^{x}}{\left(e^{x}-1\right)^{2}} d X$(2)
Debye temperature is substantially regarded as the basic parameters, and it is closely related to many properties of solids, such as melting point and specific heat and elastic constants. It is further worth noting that the Debye temperature corresponds to the highest frequency of the lattice vibrations. It is also actually a reflection of crystal combined with the strongest bond; i.e., the Debye temperature is the dividing line between high and low temperatures. This means that the high temperature is greater than the Debye temperature, which is less than the low temperature. Furthermore, the interatomic force and the Young's modulus is larger, and expansion coefficient is smaller. Therefore it can be used as a measure of the strength of the crystal covalent bond. If the Debye temperature is higher, the crystal covalent bond strength is larger [33]. In low temperatures, the Mg2Sn Debye temperature can be determined based on the elastic constant, and the average sound velocity can be obtained using the following formulas [34]:
$\Theta_{D}=\frac{h}{K_{B}}\left[\frac{3 n}{4 \pi}\left(\frac{N_{A} \rho}{M}\right)\right]^{1 / 3} V_{m}$(3)
where ΘD is the Debye temperature, h is the Planck constant, kB is the Boltzmanm constant, NAis the Avogadro number, n is the total number of atoms in the unit cell, ρ is the density and M is the molecular weight. Additionally, the average sound velocity can be calculated as follows [35]:
$v_{\mathrm{m}}=\left[\frac{1}{3}\left(\frac{2}{v_{s}^{3}}+\frac{1}{v_{1}^{3}}\right)\right]^{-1 / 3}$(4)
$v_{1}=\sqrt{\left(B+\frac{4}{3} G\right) \frac{1}{\rho}}$(5)
$v_{s}=\sqrt{G / \rho}$(6)
where νl and vs are respectively the compression velocity and shear sound velocity, B is isothermal bulk moduli, and G is shear moduli. In Eqs. (3) - (4), the Cv is constant volume specify heat. The calculated values are close to the theoretical and experimental data reported [30, 36], which considered the lattice frequency distribution. Consequently, the result is relatively close to the actual conditions. Consequently, the lattice is considered as the elastic medium to successfully explaine the situation of low-temperature heat capacity of alloy solid. Fig.3 shows that calculated and experimental values of the heat capacity at constant volume (Cv) match well at 0K and 300K. The Debye temperature (ΘD) of Mg2Sn is presented in Fig.4. Especially, this study calculated specific heat capacity (Cv) and the Debye temperature (ΘD). Consequently, the values are 14cal cell·K and 321K at 150K, and the differences are 2.6% and 1.8%. When the temperature is 300K, the Debye temperature is up to 324K, which implies that the data is closer to 277K with elastic constants when the Debye temperature is 300K.
Figure 3. The curve of constant volume specify heat
Figure 4. Calculated value of Debye temperature[19]
The trend of change is basically the same with different temperatures. Namely the trend between values of isovolumic specific heat (Cv) and Debye temperature (θD) and experimental data is substantially the same. In fact, the calculated results were slightly higher than the experimental values due to the ideal circumstances of the calculation model. However, the actual material always has grain boundary scattering, and the phonon free path must inevitably be reduced because of strongly scattering grain in boundaries, which results in the actual measured thermal conductivity being less than the theoretical value.
In summary, this work investigated the thermodynamic performances of Mg2Sn via first principles, and then discussed the thermal stability through lattice constants, band structure and elastic constants. Moreover, it demonstrated phonon density of states, constant volume and Debye temperature to study the thermodynamic properties have with calculations of the structural stability. Finally, the elastic properties and thermodynamic performances of Mg2Sn compound were mainly focused on, and Mg2Sn has demonstrated a good ability when adding alloying element Sn into Mg-alloys. The reason why experimental values are lower than the data calculated is the ideal circumstances of the Debye model and strongly scattering grain in boundaries. In addition, the calculation results show the agreement with the value of previous literatures, which means that Mg2Sn compound can be considered as candidates for the next generation of high-performance thermodynamic materials.
This work was supported by the National Youth Science Foundation of China (Grant No.11404112). This paper was also supported by the college students innovation project of North China University of Water Resources and Electric Power in 2014 and the Ministry of water resources science and technology promotion project (Grant No.TG1420).
[1] Ganeshan, S. L. Shang, Y. Wang and Z. K. Liu, “Temperature dependent dlastic coefficients of Mg2X(X=Si,Ge,Sn,Pb) compounds from first-principles calculations,” J. Alloy. Compd., vol. 498, pp. 191-198, 2010. DOI: 10.1016/j.jallcom.2010.03.153.
[2] Renbo Song, Tatsuhiko Aizawa, Atsushi Yamamoto and T. Obara, “Solid-state synthesis of Mg2Si1-xYx (Y=Ge and Sn) thermoelectric materials via bulk mechanical alloying,” Metastable and Nanocrystalline Materials, vol. 24-25, pp. 347-350, 2005. DOI: 10.4028/www. scientific.net/JMNM.
[3] J. J. Martin, “Thermal conductivity of Mg2Si, Mg2Ge and Mg2Sn,” Phys. Chem.Solids., vol. 33, pp. 1139-1148, 1972. DOI: 10.1016/S0022-3697(72)80273-7.
[4] Jun-ichi Tani and Hiroyasu Kido, “First-principles and experimental studies of impurity doping into Mg2Si,” Intermetallics, vol. 16, pp. 418–423, 2008. DOI: 10.1016/j.intermet.2007.12.001.
[5] N. Espinosa, M. Lazard, L. Aixala, et al., “Modeling a thermoelectric generator applied to diesel automotive heat recovery,” Journal of Electronic Materials, vol. 39, pp. 1446-1455, 2010. DOI: 10.1007/s11664-010-1305-2.
[6] H. Kaibe, I. Aoyama, M. Mukoujima, et al., “Development of thermoelectric generating stacked modules aiming for 15% of conversion efficiency,” Thermoelectrics, pp. 242–247, 2005. DOI: 10.1109/ICT.2005.1519929.
[7] M. El-Genk, H. Saber and T. Caillat, “Efficient segmented thermoelectric unicouples for space power applications,” J. Energy Conversion and Management, vol. 44, pp. 1755-1772, 2003. DOI: 10.1016/S0196-8904(02)00217-0.
[8] S. I. Yamaura, H. Y. Kim and H.Kimura, “Thermal stabilities and discharge capacities of melt-spun Mg-Ni based amorphous alloys,” Journal of Alloys and Compounds, vol. 339, no. 1, pp. 230-235, 2002. DOI: 10.1016/S0925-8388(01)01998-3.
[9] Linhart Ju, “Research of thermophysical and optical properties of sapphire and other high-temperature oxides with assistance of an optical furnace,” International Journal of Heat and Technology, vol. 2, no. 23, pp. 27-36, 2005.
[10] Q. M. Yang, M. Ciureanu and D. Ryan, “Composite hydride electrode materials,” Journal of Alloys and Compounds, vol. 274, no. 1, pp. 266-273, 1998. DOI: 10.1016/S0925-8388(98)00542-8.
[11] W. A. Counts, M. Friák, D. Raabe, et al., “Using AB initio calculations in designing BBC Mg-Li alloys for ultra-light weight applications,” Acta Materialia, vol. 57, no. 1, pp. 69-76, 2009. DOI: 10.1016/ j.actamat. 2008.08.037.
[12] P. Y. Tang, L. Wen, Z. F. Tong, et al., “Stacking faults in B2-structured magnesium alloys from first principles calculations,” Computational Materials Science, vol. 50, no. 11, pp. 3198-3207, 2011. DOI: 10.1016/j.commatsci.2011.06.001.
[13] P. Wang, J. P. Li, Y. C. Guo, et al., “Effect of Sn on microstructure and electrochemical properties of Mg alloy anode materials,” Rare Metal Materials and Engineering, vol. 41, no. 12, pp. 2095-2099, 2012. DOI: 10.1016/S1875-5372(13)60026-0.
[14] H. L. Gao, X. X. Liu, T. J. Zhu, S. H. Yang and X. B. Zhao, “Effect of Sb doping on the thermoelectric properties of Mg2Si0.7Sn0.3 solid solutions,” Journal of Electronic Materials, vol. 5, no. 40, pp. 830-834, 2011. DOI: 10.1007/s11664-011-1584-2.
[15] C. J. Buchenauer and M. Cardona, “Raman Scat-tering in Mg2Si, Mg2Ge and Mg2Sn,” Phys Rev B, vol. 3, no. 8, pp. 2504-2507, 1971. DOI: 10.1103/PhysRevB.3.2504.
[16] J. Tani and H. Kido, “Lattice dynamics of Mg2Si and Mg2Ge compounds from first-principles calculations,” Comp Mater Sci, vol. 42, no. 3, pp. 531-536, 2008. DOI: 10.1016/j.commatsci.2007.08.018.
[17] L. C. Davis, W. B. Whitten and G. C. Dielsin, J. Phys.Chem Solids, 28439, 1967.
[18] Kearney R. J., 1970, J. Phys .Chem. Solids, 31085.
[19] F. J. Jelinek, W. D. Shickell and B. C. Gerstein, “Thermal study of group II-IV semiconductors-II. Heat capacity of Mg2Sn in the range 5-300°K,” Journal of Physics and Chemistry of Solids, vol. 28, no. 2, pp. 267-270, 1967. DOI: 10.1016/0022-3697(67)90119-9.
[20] D. Mcwilliams and D. W. Lynch, “Infrared reflec-tivities of magnesium silicide, germanide and stannide,” Phys Rev, vol. 130, no. 6, pp. 2248-2252, 1963. DOI: 10.1103/PhysRev.130.2248.
[21] Wang Jin-Tao, Yu Wen-Li, Wang Tao, Wang Yu-Ling and Gao Yun-Liang, “First-principles study on the thermodynamic defect and crystal structure of U-12.5 At% Nb alloy,” International Journal of Heat and Technology, vol. 1, no. 33, pp. 175-180, 2015.
[22] A. K. Ganguli, A. M. Guloy and J. D. Corbett, “Concerning the Ca(2-x)MgxTt systems, Tt=Sn, Pb,” Journal of Solid State Chemistry, vol. 152, no. 2, pp. 474-477, 2000.
[23] J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., vol. 77, pp. 3865-3866, 1996.
[24] P. Ravindran, L. Fast, P. A. Korzhavyi, et al., Eriksson O., 1998, J. Appl. Phys., 84 4891.
[25] V. I. Zubov, N. P. Tretiakov, J. N. Teixeira Rabelo, et al., “Calculations of the thermal expansion, cohesive energy and thermodynamic stability of a van der waals crystal-fullerene C60,” Physics Letters A., vol. 194, no. 3, pp. 223-227, 1994. DOI: 10.1016/0375-9601(94)91288-2.
[26] O. Kubaschewsli and A. Walter, “Results of investigations of alloys by high-temperature calorimetry,” Zeitschrift fur Elcktrochemie, vol. 45, pp. 732-740, 1939.
[27] H. Zhang, S. Shang, E. S. James, et al., “Enthalpies of formation of magnesium compounds from first-principles calculations,” Intermetallics, vol. 17, no. 11, pp. 878–885, 2009. DOI: 10.1016/j.intermet.2009.03.017.
[28] A. A. Nayeb-Hashemi and J. B. Clark, “Phase diagrams of binary magnesium alloys,” ASM International, 1988.
[29] V. Tvergaard, J. W. Hutchinson, “Microcracking in ceramics induced by thermal expansion or elastic anisotropy,” J. Am. Ceram. Soc., vol. 71, no. 3, pp. 157–166, 1988.
[30] J. I. Tani and H. Kido, “Lattice dynamics of Mg2Si and Mg2Ge compounds from first-principles calculations,” Computational Materials Science, vol. 42, no. 3, pp. 531–536, 2008. DOI: 10.1016/j.commatsci.2007.08.018.
[31] O. Madelunga and B. Landolt, “Numerical data and functional relationships in science and technology,” New Series, Group III, vol. 17e, Berlin: Springer-Verlag, 1983.
[32] S. Baroni, P, Giannozzi and A. Testa, “Elastic constants of crystals from linear-response theory,” Phys. Rev. Lett., 1987. 581861. DOI: 10.1103/59.2662.
[33] Y. F. Li, Y. M. Gao, B. Xiao, et al., “Theoretical study on the stability, elasticity, hardness and electronic structures of W-C binary compounds,” Journal of Alloys and Compounds, vol. 502, no. 1, pp. 28-37, 2010. DOI: 10.1016/j.jallcom.2010.04.184.
[34] D. Music, A. Houben, R. Dronskowski and J. M. Schneider, “Ab initio study of ductility in M2AlC (M=Ti, V, Cr),” Phys. Rev. B, 75(174102), 2007. DOI: PhysRevB.75.174102
[35] J. Feng, J. C. Chen, B. Xiao, et al., “Stability, thermodynamic and mechanical properties of the compounds in the Ag–Sn–O system,” Physica B: Condensed Matter, vol. 404, no. 16, pp. 2461–2467, 2009. DOI: 10.1016/j.physb.2009.05.004.
[36] R. G. Schwartz, H. Shanks and B. C. Gerstein, “Thermal study of II–IV semiconductors: Heat capacity and thermodynamic functions of Mg2Pb from 5-300°K,” J. Solid State Chem., vol. 3, no. 4, pp. 533–540, 1971. DOI: 10.1016/0022-4596(71)90099-5.