Molecular simulation research on the micro effect mechanism of interfacial properties of nano SiO2/meta-aramid fiber
OPEN ACCESS
In this paper, the molecular dynamics simulation was used to research the micro mechanism of nano SiO2 improving the properties of poly-m-phenyleneisophthalamide (MPIA) insulation paper in oil-immersed power transformers. For the first time, the double-layer model, with silica located at the bottom layer and poly-m-phenyleneisophthalamide fibre located at the upper layer, was built in the molecular simulation software. A strong interaction between SiO2 and meta-aramid was observed, and the interaction energy was negative, which indicates that the SiO2 particles and meta-aramid fiber have formed a stable interfacial interaction. In the interaction between nano SiO2 and meta-aramid fibers, the dispersion force interaction energy was found to play a leading role in Van Der Waals force. A new hydrogen bonding network has formed between the interface of nano SiO2 and meta-aramid fiber, which is beneficial to the combination of the two. Among these models, the thermal stability and mechanical properties of the modified interface model of (3×3) are the best. The conclusions of this paper will provide a good reference for further research on the modification of nano SiO2 particles to the meta-aramid insulation paper.
Micro and Nanoscale, Interaction, Hydrogen bonds, Thermal Stability
The manufacturing requirements of aramid insulation paper is high, and Nomex paper. produced by DuPont Co., is representative of high performance aramid paper [1]. The full name of meta aramid is poly-m-phenyleneisophthalamide (PMIA), and the aramid fiber has excellent properties such as high strength, high modulus, high heat resistance and light weigh [2,3]. However, with the electirc power system reform continues to move forward [4] and the increase in the running voltage and capacity of transformers, the requirements of insulation performance and reliability of the aramid insulation paper become higher [5]. Therefore, it is urgent to carry out further research to improve the performance of aramid insulation paper.
The practice of adding nano SiO2 particles to the polymer is widely used as a physical modification method [6-9]. The mechanical properties and electrical properties of the films were studied by T. Dong [10] through hybridization of thin films by using PI/SiO2. The analysis of wide angle X diffraction showed that the introduction of SiO2 destroyed the ordered degree of the molecular arrangement of the polyimide. The tensile test showed that the tensile strength and elongation at break of the hybrid films reached the maximum when the content of spherical nano SiO2 was 3 wt%. Meanwhile, when the content of stick shaped nano SiO2 was 1 wt%, the tensile strength and elongation at break of the hybrid films reached the maximum. The modification of low density polyethylene was carried out by doping nano SiO2, and the LDPE/SiO2 was analyzed by using molecular simulation technology [11]. The results showed that the addition of 5 nm and 7 nm nanoparticles improved glass transition temperature and thermal stability.
Molecular simulation, based on the first principle [12, 13] and as a rapid developing research method, is now widely used, as Stephen W. Watt et al. [14] used molecular simulation to research the melting temperature and glass transition temperature of myo-inositol and neo-inositol. The results showed that the glass transition temperature was 217 ℃ and 245 ℃ respectively, and a small change in the molecular structure of the two molecules led to a sharp rise in the melting point. Many other scholars also used molecular simulation technology to carry out a number of research projects [15-17]. The interfacial properties of nanoparticles/polymer composites are an important index to reflect the properties of nano modified polymer materials [18-22]. Because the microscopic mechanism of interface is on the molecular level, it is difficult for traditional experimental methods to meet the experimental requirements. Molecular simulation technology can provide a new path to overcome the shortcomings of these traditional methods.
Therefore, in this paper, for the first time, the interface model of PMIA amorphous region and SiO2 is constructed by molecular simulation technology, and the interface properties of the model are studied by molecular dynamics simulation. The interaction is researched through the energy change of interface between SiO2 particles and PMIA fibers, and the microscopic mechanism of SiO2 combined with PMIA is researched by analyzing the combination of nano SiO2 and meta-aramid fiber. Furthermore, by studying the change of the mean square displacement, the effects of temperature and SiO2 lattice on the thermal stability of aramid fibers are studied. The conclusion of this paper will provide some references for further research on the modification of nano SiO2 to the aramid insulation paper
2.1 Construction of SiO2 crystal planes
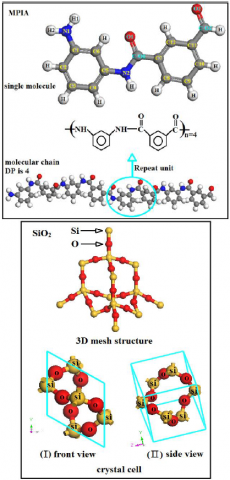
Because SiO2 (100) crystal planes contains atoms of all directions and positions, it can comprehensively analyze the interfacial interaction between SiO2 nanoparticles and PMIA. Therefore, the (100) crystal plane is selected to construct the SiO2 interface. The specific steps are as follows. Firstly, the crystal model of SiO2 is introduced. Secondly, through the Build module in the software, the (100) crystal surface is constructed, with 3 layers of SiO2, and a thickness of 1.2764nm. In order to make the cell structure more reasonable, the energy optimization of the unit cell is carried out under the COMPASS force field [23]. Then, through the Build module, super cells of (2×2), (2×3), (3×3), (4×3), (4×4) are constructed, and the 2D surface is constructed to a 3D surface through the Crystal option in the Build module.
2.2 Structure of PMIA amorphous region
Liao Ruijin et al. [24] used XRD to test the results, and used the peak separation method through Jade software to obtain the crystallinity of aramid short fiber and pulp. The results showed that the PMIA fiber structure contains an amorphous and crystalline structure, and the amorphous region accounted for 75%-80%, while the crystallization of aramid fiber itself was very low [25]. Therefore, in this paper, the model of the amorphous region of PMIA fiber is constructed, with the DP (degree of polymerization) at 4, and the model is optimized. Then, the Cell Amorphous module is used to construct a 2D amorphous structure. In the process of construction, the crystal cell parameters of the amorphous region are consistent with the lattice parameter a, b of SiO2. The molecular structure of PMIA and SiO2 is shown in Figure 1.
Figure 1. Molecular structures
2.3 Construction of interface model of PMIA amorphous region and SiO2
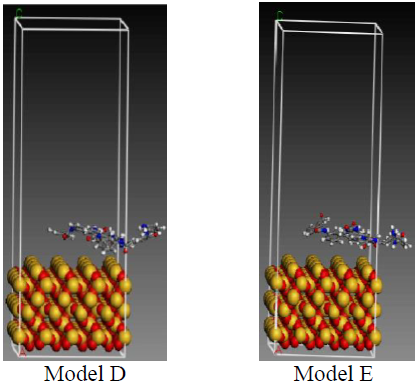
The model is constucted using the layers Build option in the Build module. The bottom layer (Layer 1) is an optimized surface model of SiO2, and the upper layer (Layer 2) is a model of the PMIA amorphous region. In order to make the aramid fiber motion unlimited in the late optimization and dynamic process, the vacuum layer of Layer 2 is set at 30. The constructed model is shown in Figure 2. In order to facilitate the description,the interface of models from small to large was recorded as model A, model B, model C, model D and model E.
Figure 2. Interfaces of the initial models
2.4 Dynamics simulation
The surface atoms are fixed before the dynamics operation in order to eliminate the influence of the surface area of nano SiO2 and the periodic boundary conditions on the result, and then the model is fully relaxed. The relaxation process is as follows. In the NVT ensemble, a cyclic relaxation process is carried out through selecting the Discover module, under the COMPASS force field, where each 100°C is a target temperature, from 30°C to 630°C, and then from 630 °C to 30°C. After each dynamics simulation, the energy of the model is minimized at each 5000 steps. After a cycle, the energy of the system tends to be stable. Each dynamics time is set to 200ps, and the time step of simulation is set to 1fs, then the molecular dynamics simulation is carried out. Because the internal insulation structure of transformers is affected by environmental factors and the electric transformation, the local hot spot temperature is taken into account. At the same time, considering the temperature of the local hot spot, the temperature of the insulation structure can reach 130 °C. Therefore, in this paper, the temperature range is selected as 70℃-150℃, and each 20℃ is a temperature point of the dynamic simulation. In the process of simulation, each model firstly carries out the dynamic simulation of 300ps in the NVT ensemble, and then in the NPT ensemble, and the molecular dynamics calculation of 300ps is carried out. The Andersen method is used for temperature control, the Berendsen method is used for pressure control with the pressure set to 1atm. Non-bond interaction is based on the atomic group's truncation method, with the truncated radius at 0.95nm, and the motion trajectory of the system is recorded every 500 steps.
3.1 Interaction of SiO2/PMIA
After molecular dynamics, the interface model of the system is shown in Figure 3. Through comparing Figure 2 and Figure 3, after molecular dynamics, significant changes have taken place in the position of the PMIA fiber molecules. In the dynamics process, the PMIA fiber molecules gradually move to the surface of SiO2, which means the interaction is relatively strong between the interface of PMIA fiber and SiO2.
Figure 3. The interfaces of models after molecular dynamics
The energy of the interface model includes non-bond interaction energy, bond interaction energy and binding energy, and the non-bond interaction energy plays a leading role in the total energy of the system. Non-bond interaction energy is composed of hydrogen bond energy, electrostatic energy and Van Der Waals force energy which includes the dispersive force energy and the repulsive force energy. The interaction energy between the interface of the PMIA amorphous region and SiO2 can be expressed by the following formula:
${{E}_{Interaction}}={{E}_{Total}}-({{E}_{PMIA}}+{{E}_{Si{{O}_{2}}}})$ (1)
where EInteraction is the interaction energy between the interface, ETotal is the total energy of the system, EPMIA is the energy of the surface of the PMIA after removing SiO2, ESiO2 is the surface energy of SiO2 after removing PMIA. The negative value of the interaction energy is the binding energy, and is expressed by formula (2), which indicates that the larger the value is, the easier it is to combine.
${{E}_{Binding}}=-{{E}_{Interaction}}$ (2)
The interaction between nanoparticles and polymer molecules leads to the formation of interfacial interactions, and this interaction is mainly reflected in Van Der Waals force, which is called Van Der Waals force energy, as shown in formula (3).
${{E}_{Vdw}}={{E}_{repulsive}}+{{E}_{dispersive}}$ (3)
Due to the limited space, here only gives the statistical average of energy of the interface structure of Models A, C and E with temperature changes, and the results are shown in Tables 1, 2 and 3.
Table 1. Statistical average of energy of Model A (1 cal = 4.1868 J)
|
kcal/ mol |
70 ℃ |
90 ℃ |
110 ℃ |
130 ℃ |
150 ℃ |
|
ETotal |
8053.58 |
8042.29 |
8034.54 |
8032.14 |
8024.35 |
|
EPMIA |
-91.67 |
-86.95 |
-81.01 |
-79.84 |
-77.57 |
|
ESiO2 |
7341.91 |
7338.19 |
7336.47 |
7335.64 |
7332.53 |
|
EVdw |
-122.37 |
-118.99 |
-123.73 |
-122.12 |
-124.50 |
|
ERevulsive |
474.58 |
153.35 |
467.43 |
153.23 |
468.93 |
|
EDispersive |
-596.95 |
-267.82 |
-591.16 |
-267.14 |
-593.43 |
|
EInteraction |
-620.00 |
-617.15 |
-617.07 |
-616.67 |
-614.25 |
|
EBinding |
620.00 |
617.15 |
617.07 |
616.67 |
614.25 |
Table 2. Statistical average of energy of Model C
|
kcal/ mol |
70 ℃ |
90 ℃ |
110 ℃ |
130 ℃ |
150 ℃ |
|
ETotal |
-18610.03 |
-18585.08 |
-18567.23 |
-18545.33 |
-18523.60 |
|
EPMIA |
-99.11 |
-91.48 |
-84.153 |
-78.25 |
-73.90 |
|
ESiO2 |
-17747.92 |
-17747.11 |
-17743.55 |
-17735.73 |
-17725.02 |
|
EVdw |
-279.88 |
-280.96 |
-285.22 |
-279.88 |
-288.58 |
|
ERevulsive |
718.31 |
716.89 |
708.40 |
716.18 |
704.46 |
|
EDispersive |
-998.18 |
-997.85 |
-993.62 |
-996.05 |
-993.04 |
|
EInteraction |
-763.01 |
-746.49 |
-739.53 |
-731.35 |
-724.68 |
|
EBinding |
763.01 |
746.49 |
739.53 |
731.35 |
724.68 |
Table 3. Statistical average of energy of Model E
|
kcal/ mol |
70 ℃ |
90 ℃ |
110 ℃ |
130 ℃ |
150 ℃ |
|
ETotal |
-31879.09 |
-31827.19 |
-31780.68 |
-31745.81 |
-31715.18 |
|
EPMIA |
-71.96 |
-60.27 |
-45.64 |
-36.38 |
-22.74 |
|
ESiO2 |
-31213.58 |
-31180.80 |
-31152.75 |
-31130.59 |
-31119.84 |
|
EVdw |
-512.18 |
-518.40 |
-516.36 |
-511.01 |
-517.12 |
|
ERevulsive |
971.03 |
957.94 |
969.54 |
971.98 |
966.58 |
|
EDispersive |
-1483.21 |
-1476.34 |
-1485.90 |
-1482.99 |
-1483.71 |
|
EInteraction |
-593.56 |
-586.13 |
-582.30 |
-578.84 |
-572.60 |
|
EBinding |
593.56 |
586.13 |
582.30 |
578.84 |
572.60 |
From the Tables 1, 2 and 3, we can see that ETotal of system are negative, which shows that the system is stable. The energy values are close to each other, and the fluctuation range is small with an increase in the temperature, which indicates that the influence of temperature on the interfacial interaction between the PMIA amorphous region and the SiO2 is small. With an increase in temperature, each energy value increased in addition to the binding energy. This is because the kinetic energy increased with the increase in temperature. Furthermore, due to lower temperature, the combination was closer, and the interaction was stronger. So, the smaller the negative value of the interaction energy was, the greater the binding energy was. On the contrary, the higher the temperature was, the greater the kinetic energy of the molecule was, the less the combination degree was, the weaker the interaction was, and the less the interaction energy was, the smaller the binding energy was. Throughout the system, because the number of SiO2 atoms is large and the number of PMIA is small, the proportion of ESiO2 is larger, and it has a greater influence on ETotal. ERevulsive is positive, and EDispersive is negative, so the dispersive force energy plays a lead role in Van Der Waals force energy.
In order to study the effect of interface size on the interfacial characteristics of the PMIA amorphous region and SiO2 under the same temperature, 90℃ is used as an example, and the results are shown in Table 4.
Table 4. Statistical average of energy of different models at 90℃
|
kcal/ mol |
Model A |
Model B |
Model C |
Model D |
Model E |
|
ETotal |
-8042.293 |
-12576.325 |
-18585.082 |
-31126.845 |
-31827.188 |
|
EPMIA |
-86.951 |
-83.532 |
-91.476 |
-79.656 |
-60.266 |
|
ESiO2 |
-7338.188 |
-11862.377 |
-17747.112 |
-30408.283 |
-31180.795 |
|
EVdw |
-118.991 |
-192.837 |
-280.962 |
-483.654 |
-518.402 |
|
ERevulsive |
153.346 |
553.862 |
716.891 |
914.309 |
957.942 |
|
EDispersive |
-267.823 |
-746.699 |
-997.853 |
-1397.963 |
-1476.344 |
|
EInteraction |
-617.154 |
-630.416 |
-746.4942 |
-638.906 |
-586.126 |
|
EBinding |
617.1541 |
630.416 |
746.4942 |
638.9058 |
586.1261 |
In order to observe the changes of energy with the change of the size of lattice, according to the results of Table 4, Figure 4 and Fig. 5 are drawn:
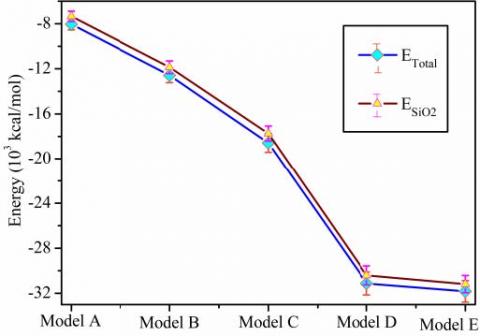
Figure 4. ETotal and ESiO2 change with the size of lattice
Figure 4 shows that the trend of ETotal and ESiO2 is the same, and with an increase in the lattice size of SiO2, the two increase along the negative direction. At 90℃, the superlattice of Model E has increased by about 295.75% over that of the superlattice of Model A along the negative direction.
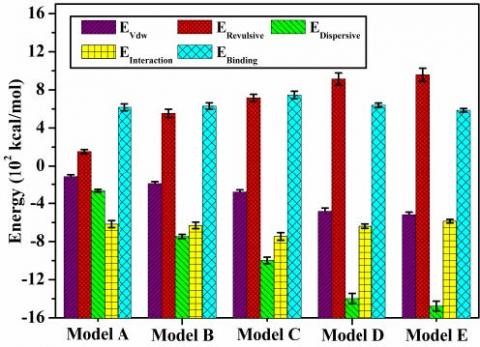
Table 4 and Figure 5 show that with an increase in lattice size, EVdw, EDispersive and EInteraction all have a significant increase trend along the negative direction. The dispersive force effect is the dominant effect on the interfacial interaction between the PMIA amorphous region and SiO2 of non-bond interaction. With an increase in the lattice size, the interfacial interaction energy increases along the negative direction, which indicates that the interface between PMIA and SiO2 is enhanced, and it is beneficial for the nano SiO2 particles doping into PMIA fibers. Overall, the interfacial interaction energy is negative, the binding energy is positive, and with an increase in SiO2 surface, the binding energy of the two has an increasing tendency, which is beneficial for the stable combination of SiO2 particles and PMIA fibers. Moreover, the interfacial interaction between MPIA molecules and nano SiO2 is related to the size of the interface. The binding energy of Model C is the largest, and its interfacial combination effect is the best.
Figure 5. EVdw, EDispersive, EInteraction, EInteraction, EBindingof interface change with the lattice
3.2 Hydrogen bonds
The hydrogen bond network formed between molecules of polymer is beneficial to the stability of the molecular structure, and determines the structure of the molecule to a certain extent, such as melting point, boiling point, etc. [26].
A strong non-bond interaction formed between H atoms with large electronegativity and adjacent atoms with large electronegativity is referred to as a hydrogen bond. Its definition is divided into the geometric principle and the energy criterion [27]. The definition of hydrogen bond is used in this paper.
Figure 6. Hydrogen bonds network, 1 represents the MPIA intramolecular hydrogen bonds, 2 represents the intermolecular hydrogen bonds
The maximum distance of acceptor atoms and hydrogen atoms is 3 Å, the minimum value of the angle of donor atoms, hydrogen atoms and acceptor atoms is 90。.
The interface Model A is used as an example of the hydrogen bonding. Through the dynamics, the hydrogen bonds of the model of aramid fiber and SiO2 are shown in Figure 6. In order make the figure clearer, the background color is black.
As shown in Figure 6, the intramolecular hydrogen bonds are formed in the N, H and O atoms of aramid fiber, and the interfacial hydrogen bonds are formed between the hydrogen bonds H atoms of aramid fiber and O atoms of SiO2. Compared to the interfacial hydrogen bonds, the number of intramolecular hydrogen bonds in aramid fiber is large. This is because the spacing of H atoms to O and N atoms is small, which easily meets the necessary conditions for the formation of hydrogen bonds, thus making the probability of forming hydrogen bonds high. The hydrogen bonding between aramid fiber and SiO2 is beneficial to the bonding between aramid fiber and SiO2.
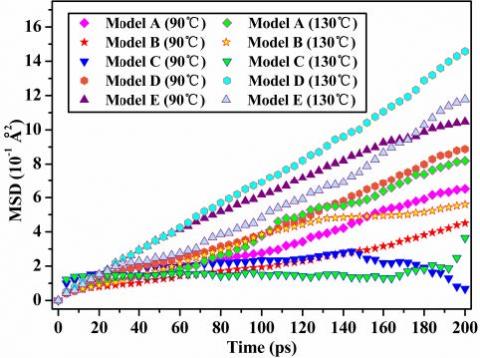
Figure 7. The MSD of different models at different temperatures
The relationship between the time and the mean square displacement (MSD) is expressed by the formula (4) [28], where ${{\vec{r}}_{i}}(t)$ represents the coordinate of ith molecule at time t of the system, ${{\vec{r}}_{i}}(0)$ represents the initial coordinate of this molecule, which can be used to reflect the chain motion of polymer. The greater the slope of MSD and time curve, the more intense the chain motion of polymer, and the worse the stability of polymer.
$MSD=\left\langle |{{{\vec{r}}}_{i}}(t)-{{{\vec{r}}}_{i}}(0){{|}^{2}} \right\rangle $ (4)
The MSD of the amorphous region of PMIA changes with the temperature, as shown in Figure 7.
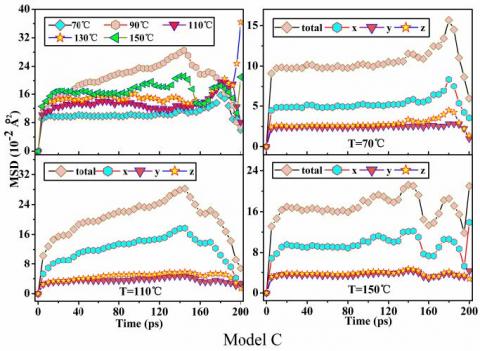
Figure 7 shows that the higher the temperature, the greater the value of PMIA, and the stronger MSD chain motion, the greater the average kinetic energy of PMIA, which is consistent with the relationship between temperature and the average kinetic energy of molecules. This fully shows that an increase in temperature causes a decrease in thermal stability of PMIA. In the initial stage of the curve (<80ps), except for Model D (130 ℃) and Model E (90 ℃), the MSD of the other models has little difference. After 80ps, on the whole, the MSD of Model D reaches the maximum, and the MSD of Model C reaches the minimum. The chain motion of Model C is the weakest, which indicates that the SiO2 of Model C has a stronger binding effect on MPIA molecules, which indicates the thermal stability of Model C is the best among the interface models of this paper.
In Figure 7, the majority of the curve has a good linear relationship with time, and the curve end is irregular due to the statistical error. The mean square displacement of MPIA molecules along the X direction are obviously greater than that of the other two directions, which indicates that the chain motion of surface along the X direction is relatively strong. Furthermore, the X direction being in the direction of interface formation indicates that the interface interaction of MPIA molecules and SiO2 is relatively strong.
In this paper, molecular dynamics simulations are carried out to study the interfacial interaction in micro and nanoscale between the amorphous region of PMIA and SiO2, and the characteristics of the interface between the two and the effect of the lattice size on the thermal stability of PMIA are studied. Firstly, through the visual interface of MS software, a model of PMIA and SiO2 is constructed. Then through the relaxation process, the model is energy optimized. Finally, the molecular dynamics simulation is carried out. Through the relationship between the interfacial energy and lattice, and the analysis of hydrogen bond interaction and the mean square displacement, the following conclusions are obtained.
1) A stable interface can be formed between MPIA molecules and SiO2, with the dispersion force as the main reason for the formation of the interface between the two. The binding energy of Model C is the largest, and its interfacial combination effect is the best.
2) In the bonding process of nano SiO2 and aramid fiber, the hydrogen bond is formed between the H atoms of the aramid fiber and the O atoms of SiO2, which is beneficial for the combination of aramid fiber and SiO2.
3) With an increase in temperature, the chain motion of PMIA increases, and the thermal stability of PMIA decreases. Among the interface models in this paper, the thermal stability of Model C is the best.
This paper provides a solid reference for further research on the modification of nano SiO2 particles to the meta-aramid insulation paper.
The authors wish to thank the scientific project funded by the State Grid Chongqing Electric Power Co. Chongqing Electric Power Research Institute, and the “Fundamental Research Funds for the Central Universities” (XDJK2014B031 and XDJK2016D017) for their financial support.
[1] Zhao H.F., Zhang M. Y., Lu J. B. (2010). Configuration of PMIA-pulp and its effect on aramid paper, China Pulp & Paper, Vol. 29, No. 2, pp. 1-5.
[2] Zhang S.F. (2010). Correlation between the interface and structure characteristics of meta-aramid fiber and the properties for sheetmaking, Ph.D. dissertation, School of Papermaking Engineering, Shaanxi University of Science & Technology, Xi An, China, 2010.
[3] Jenkins M.J., Harrison K.L. (2006). The effect of molecular weight on the crystallization kinetics of polycaprolactone, Polymer for Advanced Technologies, Vol. 17, No. 6, pp. 474-478. DOI: 10.1002/pat.733
[4] Qiu W., Zhang C., Guo D., Xu Y., Wang G. (2014). A smart grid client-side testing platform for monitoring, Archives of Mechanical, Electrical and Civil Engineering, Vol. 1, No. 1, pp. 9-12. DOI: 10.18280/amece.010102
[5] Yang L., Liao R., Sun C., Sun H. (2010). Influence of natural ester on thermal aging characteristics of oil-paper in power transformers, European Transactions on Electrical Power, Vol. 20, No. 8, pp. 1223-1236. DOI: 10.1002/etep.398
[6] Li H. Y., Wu Q. L., Zhou D. G. (2015). Preparation and property analysis of cellulose nano-fibril and nano-silicon dioxide composites, Transactions of the Chinese Society of Agriculture Engineering, Vol. 31, No. 7, pp. 299-303.
[7] Li X., Cao Z., Zhang Z., Dang H. (2006). Surface-modification in situ of nano-SiO2 and its structure and tribological properties, Applied Surface Science, Vol. 252, pp. 7856-7861. DOI: 10.1016/j.apsusc.2005.09.068
[8] Li Z., Zhu Y. (2003). Surface-modification of SiO2 nanoparticles with oleic acid, Applied Surface Science, Vol. 211, pp. 315-320. DOI: 10.1016/S0169-4332(03)00259-9
[9] Cheng B., Wang X., Wang L., Wu Y. (2007). Surface modification of sub-micron spherical SiO2 particles with butanedioic acid for electrophoresis in tetrachloroethylene, Materials Letters, Vol. 61, pp. 1350-1353. DOI: 10.1016/j.matlet.2006.07.066
[10] Dong T.Q. (2007). Investigation on mechanical properties and electric properties of polyimide/silica nano-hybrid films, M.S. thesis, School of materials science and Engineering, Harbin University of Science and Technology, Harbin, China.
[11] Fan P. (2015). Research on space charge properties of LDPE/SiO2 nano-composite under thermal aging conditions, M.S. thesis, School of Electrical Engineering, Chongqing University, China.
[12] Li Y., Zhang Y., Kong X., Deng Y., Zhang R., Tang J. (2016). Investigation on thermodynatic performances of Mg2Sn compound via first principle calculations, International Journal of Heat and Technology, Vol. 34, No. 1, pp. 110-114. DOI: 10.18280/ijht.340116
[13] Li Y., Zhang Y., Kong X., Ding Y., Zhang R., Tang J. (2016). Thermal stability of the Mg2Ni-based hydrogen storage alloy doped Ti element, International Journal of Heat and Technology, Vol. 34, No. 2, pp.245-250. DOI: 10.18280/ijht.340213
[14] Watt S.W., Chisholm J.A., Jones W., Motherwell S. (2004). A molecular dynamics simulation of the melting points and glass transition temperatures of myo- and neo-inositol, The Journal of Chemical Physics, Vol. 121, No. 19, pp. 9565-9573. DOI: 10.1063/1.1806792
[15] Varanasi S.R., Guskova O.A., John A., Sommer J.–U. (2015). Water around fullerene shape amphiphiles: A molecular dynamics simulation study of hydrophobic hydration, The Journal of Chemical Physics, Vol. 142, No. 224308, pp. 1-15. DOI: 10.1063/1.4922322
[16] Hori T., Shiga T., Shiomi J. (2013). Phonon transport analysis of silicon germanium alloys using molecular dynamics simulations, Journal of Applied Physics, Vol. 113, No. 203514, pp. 1-6. DOI: 10.1063/1.4807301
[17] Yang Z., Zhao J., Wei N. (2015). Temperature-dependent mechanical properties of monolayer black phosphorus by molecular dynamics simulations, Applied Physics Letters, Vol. 107, No. 023107, pp. 1-5. DOI: 10.1063/1.4926929
[18] Mei Q., Li C., Wang J., Chen J.F., Le Y. (2014). Molecular dynamics simulation on the interaction of CeO2 and silane coupling agent in solutions, Materials Research Bulletin, Vol. 49, pp. 265-271. DOI: 10.1016/j.materresbull.2013.09.001
[19] Li X., Wang G., Li X., Lu D. (2004). Surface properties of polyaniline/nano-TiO2 composites, Applied Surface Science, Vol. 229, pp. 395-401. DOI: 10.1016/j.apsusc.2004.02.022
[20] Cai Z., Zeng D., Liu J. (2005). The Influence of truncated radium on the molecular dynamics simulation of the interface between coexisting phases, 5th, International Symposium on Multiphase flow, Heat Transfer and Energy Conversion, Xi An, China.
[21] Yang Y. (2007). The study of molecular simulation on the interface of carbon giber/epoxy resin, M.S. thesis, Department of Applied Chemistry, Harbin Institute of Technology, Harbin, China.
[22] Sui S. Research on the influence of interface on dielectric properties of epoxy nanocomposites, M.S. thesis, School of materials science and Engineering, Harbin University of Science and Technology, Harbin, China, 2014.
[23] Xie J., Tang C., Li X., Zhou Q., Xie J., Hu D. (2016). Force field properties in molecular simulation of amorphous region in cellulose insulation paper, Oxidation Communications, Vol. 39, No. 1 A, pp. 1236-1246.
[24] Liao R., Li X., Yang L., Bai G. (2015). Effect of the ratios of aramid fiber to pulp on the properties of aramid paper, High Voltage Engineering, Vol. 41, No. 2, pp. 364-373. DOI: 10.13336/j.1003-6520.hve.2015.02.002
[25] Jain A., Vijayan K. (2002). Thermally induced strrctural changes in Nomex fibers, Bulletin of Materials Science, Vol. 25, No. 4, pp. 341-346. DOI: 10.1007/BF02704129
[26] Zhang S., Tang C., Chen G., Zhou Q., Lv C., Li X. (2015). The influence and mechanism of nano Al2O3 to the thermal stability of cellulose paper, Sci Sin Tech, Vol. 45, No. 11, pp. 1167-1179. DOI: 10.1360/N092015-00207
[27] Chen C., Li W. (2009). Molecular dynamics simulation of hydrogen bonding characteristics in aqueous glycerol solutions, Acta Physico-Chimica Sinica, Vol. 25, No. 3, pp. 507-512.
[28] Hofmann D., Fritz L., Ulbrich J., Paul D. (2000). Molecular simulation of small molecule diffusion and solution in dense amorphous polysiloxanes and polyimides, Computational and Theoretical Polymer Science, Vol. 10, pp. 419-436. DOI: 10.1016/S1089-3156(00)00007-6